
RAB32, a new Parkinson's gene
A new breakthrough in Parkinson's genetics

Happy Sunday! A new gene, RAB32, has been linked with Parkinson's disease (PD) through exome sequencing of PD families. Two groups have independently identified the gene through analyses of independent cohorts of familial PD (Hop, Lai, et al. Nat Genet 2024; Gustavsson et al. Lancet Neurol 2024). Mind you, RAB32 is not just another gene that is getting linked to PD. It comes as an important piece in the puzzle researchers have been putting together for decades since LRRK2, a frequently mutated gene in PD, was discovered.

I've known about LRRK2 and that many companies are developing drugs to inhibit this gene in the brain to treat PD. But I never realized LRRK2 is a field of its own and hundreds of researchers across the world are religiously studying this one gene since 2004, trying to understand how LRRK2 mutations result in the death of dopaminergic neurons in the brain. It all started with two publications in 2004 in the journal Neuron.
One of the many loci mapped to PD through family-based linkage analysis was PARK8, a large locus spanning the centromere ends of short and long arms of chromosome 12, reported first in 2002. The linkage region spans many millions of base pairs and hold more than 100 genes. The locus was originally mapped in a Japanese family, but soon in families in the Europe, UK and US. The PD-causing gene was dragged out of the gene-dense forest in 2004 by two research groups, one led by Thomas Gasser from the University of Tübingen in Germany (Zimprich et al. Neuron 2004) and the other, Andrew Singleton from the NIH in the US (Paisán-Ruíz et al. Neuron 2004)

Thanks to a series of families from Basque region of Spain, the disease markers that segregated in these families helped Singleton and colleagues to narrow down the disease locus from ~12 Mbp containing 116 genes to ~2 Mbp containing 11 genes and eventually to the causative gene, LRRK2. The gene encodes a gigantic protein made of 2,482 amino acids. Honoring the Basque families that helped clone this gene, the authors named this gene as dardarin, based on the Basque word 'dardara', meaning tremor.
It was the first time the cell signaling field was hearing about this gene. The researchers didn't know what kind of protein LRRK2 was making. Based on the amino acid sequence, they noticed that there were many domains, one of which was a kinase domain where many of the disease mutations appeared to cluster. The gene encoded a kinase protein (that is, it is capable of enzymatically adding a phosphate group to another protein or to itself), and the kinase activity is perhaps at the center of the biology of LRRK2-mediated PD, the researchers guessed. They proved the same in two years. Removing the kinase domain of LRRK2 eliminated its neurotoxicity. So, inhibiting the kinase activity of LRRK2 is the key to treating PD, at least the one caused by LRRK2 mutations. This realization marked the beginning of the quest that many researchers, particularly the ones in the cell signaling research, would dedicate their careers to understanding the molecular mechanisms underlying the link between LRRK2 and PD.

On one side, drug companies focussed on developing safe, CNS-penetrant LRRK2 inhibitors to treat PD. Denali therapeutics is at the forefront of this area; its drug candidate is currently in late stage clinical trials. On the other side, cell signaling researchers focussed on tracing the signaling pathways that link LRRK2 to neurodegeneration. I learned much of this from listening to the lectures (2017, 2020, 2021) of Dario Alessi, renowned PD researcher from the University of Dundee in the UK. Before 2004, Alessi was working on the protein kinase biology in the disease areas like hypertension and cancer. But then the news about LRRK2 hit Alessi like a calling and as a result, he spent the next 20 years studying LRRK2.
In the early years of LRRK2 research, Alessi and others in the field were trying to answer one question. Every protein kinase must have a substrate. What is the physiological substrate of LRKK2? This seemingly simple question costed more than a decade for the researchers to arrive at the answer. Through mass spectrometry experiments, studying the phosphorylation of profile of hundreds of proteins in the presence and absence of LRKK2 activation in cell cultures and in the brain tissue of animal models, researchers found in 2016 that the substrate of LRRK2 is a family of proteins called Rab (Ras-related protein in brain).

Rab proteins are GTPases, and there are 70 Rab GTPases known to exist in the humans, performing a wide array of molecular functions. A subset of these interact with LRRK2 and contribute to neurodegeneration. Researchers have short-listed a few that are phosphorylated by LRRK2 such as Rab8, Rab10 etc. and studying those in detail will help understand how their phosphorylation leads to neuronal death. Phosphorylation of these Rab GTPases seem to impact a wide range of cellular processes such as protein trafficking, ciliogenesis, etc. Understanding which of these are in the causal pathways to neurodegeneration is critical to inform therapeutic development. Notably, none of these downstream candidate Rab GTPases have been linked to PD via human genetics.
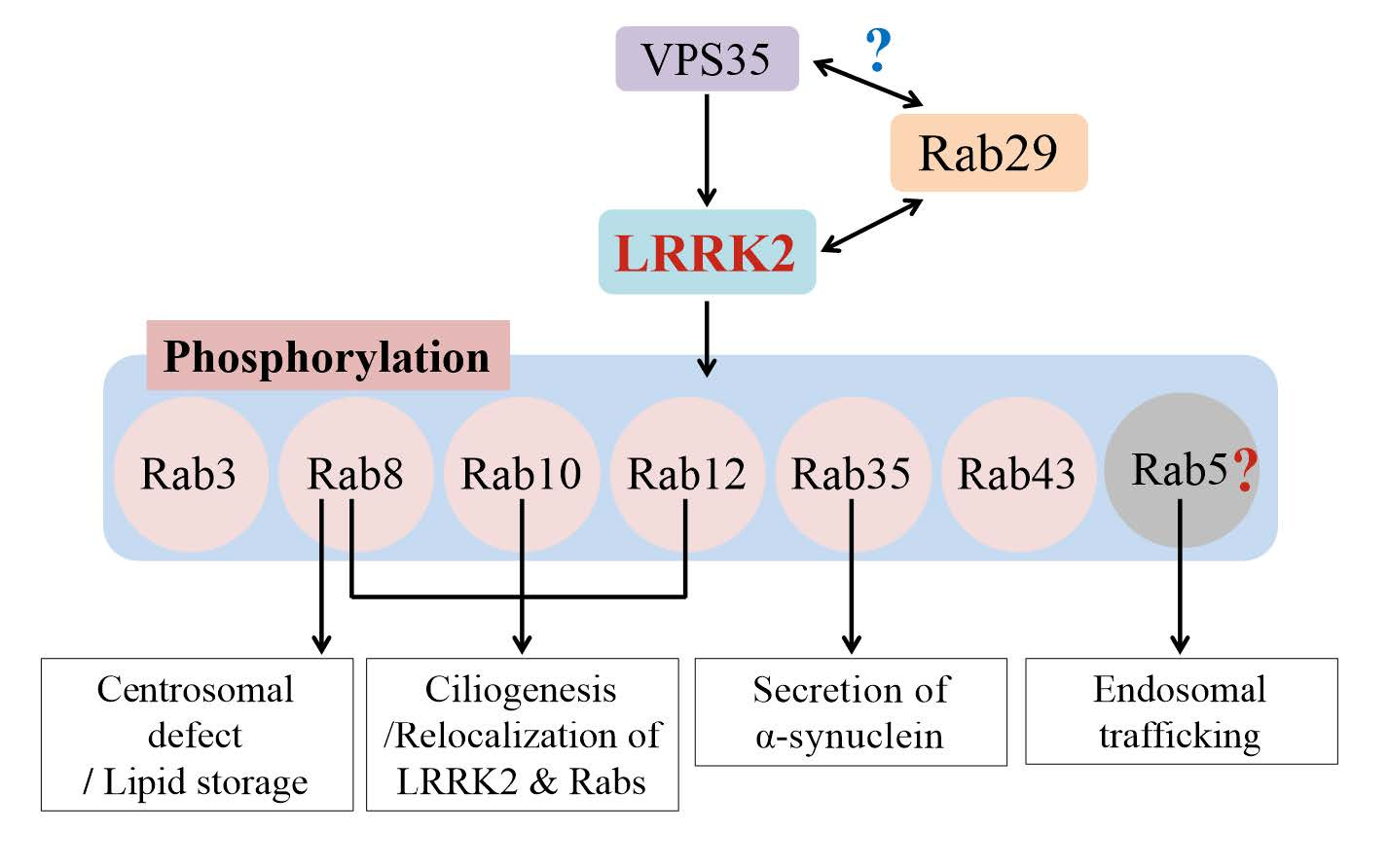
After figuring out the physiological substrates of LRRK2, researchers wondered what's upstream of LRRK2. It turns out LRRK2 is mainly a cytosolic protein, but Rabs are membrane-bound proteins. The Rabs are bound to the plasma membrane but also to the membranes of cell organelles like ribosomes, lysosomes, endoplasmic reticulum and Golgi. So, something has to reel the LRRK2 proteins from cytosol to the membranes where Rabs hang out. When the LRRK2 are brought to the membranes in proximity to the Rab proteins, it gets activated. So came the next big question: what recruits LRRK2 to its site of action? Researchers feel that the answer to this question will help understand the molecular mechanisms of LRRK2 activation, which in turn will help design safe and effective drugs to inhibit LRRK2 activation to treat PD. So far, two proteins, Rab29 and VPS35, were found to contribute to LRRK2 activation from upstream.
The RAB29 gene (also known as RAB7L1) is located in the PARK16 locus (one of the loci discovered by early GWAS studies) and is believed to be the causal gene, though no strong rare variant associations seem to exist in the literature, except for a few candidate gene studies. However, Rab29 protein has been extensively studied, particularly by Alessi's research team, in the context of LRRK2 activation. Rab29 binds to LRRK2 through one of LRRK2's non-catalytic domains called armadillo domain. Below is a beautiful electron microscope-based structures of LRRK2 bound to Rab29 in different configurations (monomeric, dimeric and tetrameric) reported in Science last year.

The other protein that is shown to activate LRRK2 from upstream is VPS35, which encodes a component of a multi-protein complex called retromer complex that transports used and worn out proteins by transporting them from endosomes to the Golgi for recycling. Mutations in VPS35 causes autosomal dominant Parkinson's disease, and it turned out LRRK2 is over-activated when VPS35 is mutated. The VPS35 protein seem to negatively impact LRKK2 activation. Monocytes and neutrophils of PD patients with VPS35 mutations show excessive phosphorylation of Rab10 (LRRK2’s substrate), suggesting LRRK2 may be mediating the neurodegeneration in PD patients with VPS35 mutations. The exact mechanism through which VPS35 contribute to LRRK2 activation is yet to be understood.
With the recent ExWAS of familial PD, Rab32 now joins the Rab party. Studying the exome sequences of 2,184 familial PD cases and 69,775 controls, an international research team discovered a missense variant, Ser71Arg, in RAB32 that increases the risk of PD by almost the same effect size as some of the severe LRRK2 risk variants. And it is seen in 0.7% of the familial PD cases. The gene also came up as the top risk gene in an independent study that focussed specifically on the Rab mutations in a small sample of familial PD cases. Probably, we will read more reports replicating RAB32's link with PD in the near future as many will now scan through the exomes of their PD cohorts, looking for Ser71Arg mutation.

Rab32 is already known to directly interact with LRRK2 through its armadillo domain, just like Rab29. The two studies that report the RAB32 mutation also show some evidence that LRRK2 and its downstream Rab10 are both hyperphosphorylated in the blood cells of patients with RAB32 mutation. Interestingly, the missense variant position 71 is the only phosphorylation site in RAB32. It's not clear if Rab32 is upstream or downstream of LRRK2 in the PD pathology (looks like upstream). But we will know soon, as many in the LRRK2 research have probably already started investigating RAB32.
The Ser71Arg mutation appears to be a gain of function mutation. Many questions to answer. Will Rab32 inhibition help treat PD in these patients? If Rab32 plays a key role in LRRK2 activation, then, will PD patients with LRRK2 mutations benefit from Rab32 inhibition? Conversely, will PD patients with RAB32 mutation will benefit from LRRK2 inhibition. This would expand the patient population for LRRK2 inhibitors. Clearly, the discovery of Rab32’s link with PD has opened many exciting research questions to explore.
