
Solving a 25-year-old genetic puzzle
The genetic mutation behind spinocerebellar ataxia type 4, first described in the 1990s, finally comes to light

I hope your week is off to a great start! The past few days, I’ve been digging into the Spinocerebellar ataxia type 4 (SCA4) literature, inspired by a new Nature Genetics paper on the successful mapping of the causative gene and mutation underlying SCA4. I've read this work last year when it was preprinted and tweeted about it. I've mentioned this preprint in a couple of my past substack posts in relation to long-read sequencing, and discussed it in the Genetics podcast with Patrick Short. But rereading the paper in its final published form helped me see that there are more interesting things buried in the SCA4 literature, which I didn't notice before.

My interest in the repeat expansion-related neurodegenerative diseases has been growing recently, as there are currently many active efforts in the field to develop therapeutics for these conditions. With the recent advancements such as long-read sequencing, single-cell sequencing etc., researchers are beginning to understand the molecular mechanisms underlying neurodegeneration (for example, cell type specific somatic expansions of repeats and how they kill the striatal neurons in Huntington's disease) and ways to prevent them (for example, targeting DNA repair genes to halt the pathological repeat expansion). So, I might be digging more into the repeat-expansion literature in the near future.
The fact about SCA4 that caught my attention was that its cause remained a mystery for more than 25 years due to high complexity of the SCA4 locus (16q22.1), and then long-read sequencing technology helped scientists to identify the disease gene (ZFHX3) and mutation (expanded GGC repeats). But there is more to the story.

At least four independent groups have successfully mapped the GGC repeat expansion in ZFHX3 to SCA4: one group from the University of Utah (where the index family was first documented in 1994, the linkage to 16q22.1 was mapped in 1996, and the ataxia was officially labelled as type 4), two groups from Sweden (one from the Lund University and the other from Karolinska Institute) where the pathogenic mutation was believed to be born in some family in Southern Sweden, possibly in the early 19th century. And the last group of researchers were from University College London and University of California.
Amazing, isn't it? After the initial linkage report in 1996, the SCA4 disease remained unsolved for decades and then boom!, suddenly four research groups (maybe there’s more, who knows) are deciphering the mystery. The interesting fact is not all the groups solved the case using long-read sequencing; two groups caught the GGC repeat just using short-reads. Particularly, the Swedish researchers had the advantage of studying multiple families descended from the founder, thereby able to narrow down the disease region using identity by descent (IBD) analysis from an initial 1.64 Mbp segment to a final 111 Kbp segment covering the very last exon of the ZFHX3 gene containing the microsatellite. Note, the ZFHX3 is a huge gene, encoding a protein of length 3703 amino acids; the start and end coordinates span more than 1 Mbp, and there are 760 naturally occurring microsatellites spread across the gene! Narrowing the disease locus using IBD analysis helped the Swedish team to zero in on the culprit. However, long-read sequencing played an important role in all the four research groups' work. It helped validate the repeat expansion and appreciate an important difference (apart from the length) in the repeat sequences between cases and controls. While the normal length repeats (between 20-26) were randomly interrupted by either synonymous or non-synonymous SNVs, expanded pathogenic repeats (>45) had no such interruptions at all. It's just GGC, GGC, GGC all the way.
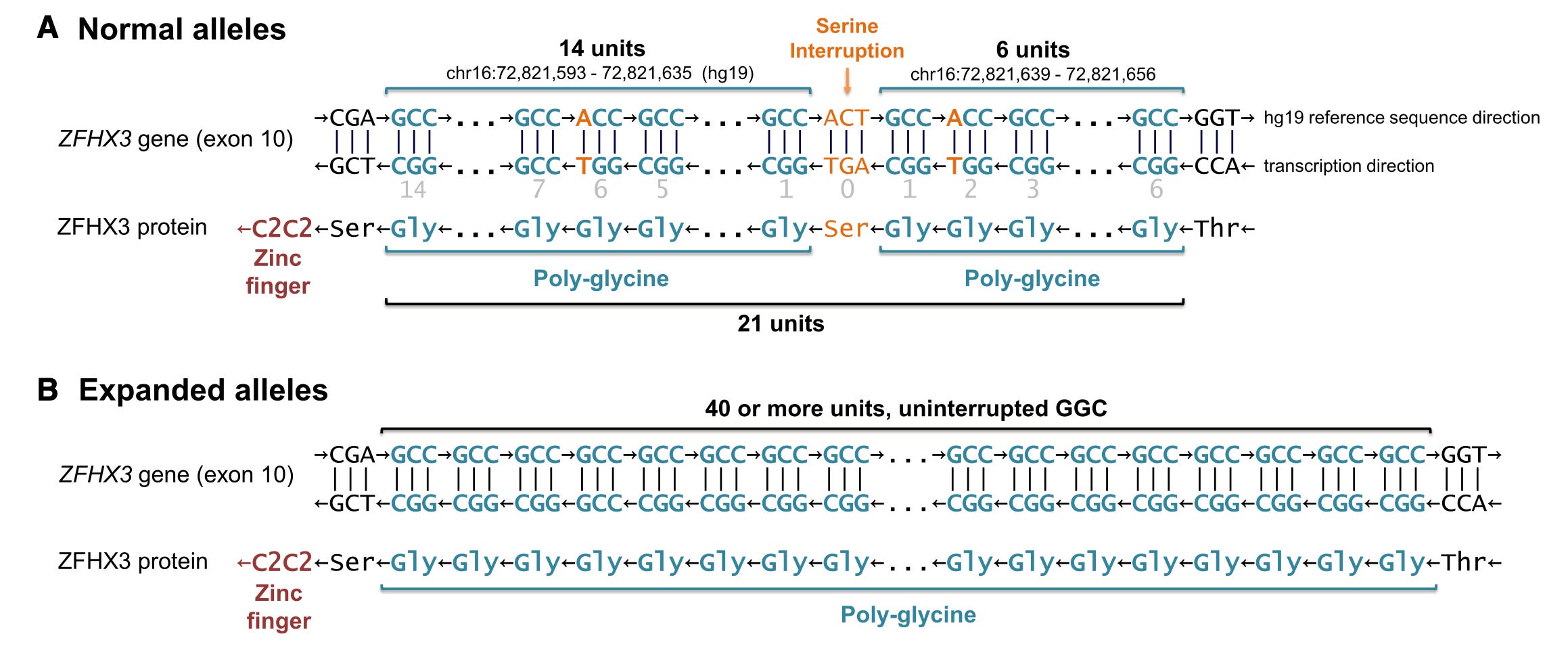
The SNV interruptions is what seem to be preventing this microsatellite from ballooning generation after generation in the general population. This makes me wonder if simply breaking the GGC monotony in the genomes of cerebellar neurons by some means, perhaps, gene editing one or two single base pairs, will help halt the disease progression. We are nowhere near gene editing the neurons in the brain, but if in the near future CRISPR or prime editors hit the brain, would such an approach help? I guess, it will make sense only if the disease pathology is due to somatic expansions of the GGC repeat, as seen in Huntington's. I don't think anyone has looked into it so far. Hopefully, someone will eventually do.
The other fascinating thing about SCA4 is its locus—16q22. This genomic region is extraordinarily complex, filled with many microsatellites, segmental duplications, pseudogenes etc. That is why it was challenging to discover the causative gene and mutation initially. The research group from UCL (Chen and Gustavsson et al.) writes in their report in Movement Disorders, "we found that the 16q22.1 region harbors the largest number of naturally occurring STRs (short tandem repeats) and naturally occurring GGC repeats compared to all other chromosomal regions, when normalized for size". Below is the density of STRs in 16q22.1 compared against rest of the genome.

It turns out that ZFHX3 is not the only ataxia gene sitting in the 16q22.1 region. At least two other ataxia genes were identified in this locus. After the initial linkage report of 16q22.1 locus to SCA4 in 1996, there were similar linkage reports in ataxia families in Southern Sweden, Northern Germany, Japan, China and India, all mapped to chromosome 16q22. The ataxia in the Japanese families was found to be caused by pentanucleotide repeat expansion in BEAN1, and the ataxia in Chinese families was found to be caused by CAG repeat (the classic polyglutamine repeat) in THAP11. The families in the Sweden were affected by the same mutation (GGC repeat in SFHX3) as the index family from Utah (who were actually immigrants from Sweden). The ataxia in the German families was phenotypically similar to SCA4, but I am not sure if the causative mutation was identified yet. If so, it would indicate an independent origin of ZFHX3 mutation, because the rest of all the SCA4 families appear to descend from a single founder, who may have lived in Southern Sweden sometime around the early 19th century. So far, it looks like SCA4 is private to this Swedish extended pedigree, who has been branching their family tree for over 200 years. Cerebellar ataxia with sensory involvement in a patient with Scandinavian ancestry, GGC repeat in ZFHX3 should be first suspect.
At the molecular level, the cerebellar neurons from postmortem tissue of deceased SCA4 patients show intranuclear inclusion bodies. This makes SCA4 officially a member of intranuclear inclusion diseases caused by repeat-expansions, which include
- Neuronal intranuclear inclusion disease (NIID) (caused by GGC repeat in NOTCHNLC),
- Fragile X-associated tremor-ataxia syndrome (FXTAS) (caused by CGG repeat expansion in FMR1 in the 'premutation' range, typically 55–200 repeats)
- Oculopharyngeal muscular dystrophy (OPMD) (caused by GCG repeat expansion in PABPN1)
The important characteristic that differentiates SCA4 from these intranuclear inclusion diseases is the relatively smaller repeat size and location of the repeat within an exon.
In the Nature Genetics paper, the research team from Utah (Figueroa, Gross, Buena-Atienza, et al.) report, in addition to intranuclear inclusions, the neurons from SCA4 patients also show signs of abnormal autophagy reminiscent of that seen in the well recognized SCA2 (caused by CAG repeats in ATXN2) and TDP-43 proteinopathies (ALS and frontotemporal dementia). In SCA4 patient-derived fibroblasts and iPSCs, the authors demonstrate cellular markers of reduced autophagy, including elevated wild-type ATXN2 (which is known to directly interact with proteins involved in autophagy). ATXN2 is already being explored as a common target for SCA2 and ALS. ATXN2 antisense oligonucleotides (ASOs) are already being tested in clinical trials for the treatment of ALS. The authors speculate if the SCA4 could be another indication for ATXN2 ASOs.
Overall, I find the SCA4 story extremely fascinating. An unlucky Swede from the early 19th century born with a pathogenic mutation in ZFHX3 has left a legacy of a mysterious brain illness that will haunt many of his/her descendants for the next 200 years and even more. Fate would have it that one of those families will leave Sweden, travel across the world and settle in Utah in the United States, revealing to the local neurologists their brain disease in the 1990s. Their DNA and the blood cell lines would sit on the lab shelves in the University of Utah, hiding a secret that would not come to light for another 25 years. Who knows how many more years it will take for a treatment to emerge that will finally undo the curse of this unfortunate Swedish pedigree.

As someone with OPMD, I found your article fascinating. Thank you!