
February 2024 human genetics roundup
A brief look at the most exciting stories from Feb, 2024


Happy March! I managed to put together another roundup post covering some studies published in February that I liked. Overall, it has been an interesting month filled with many events that fueled entertainment, heated discussions, outcries and memes in the scientific Twitter.
A review article in a Frontiers Journal was shredded to pieces by the scientific community after it was found that the authors used AI to generate nonsensical illustrations that got past the editor's and reviewers' notice.
The flagship paper of All of Us biobank was published in Nature, announcing the release of ~250,000 whole genome sequences of a diverse set of participants. The paper became popular but for the wrong reasons. If there were people in the scientific community who didn't know before what is "UMAP", there is a good chance they know now. A UMAP-based representation of the genetic diversity of the All of Us biobank went viral in a bad way after Jonathan Pritchard, a population geneticist from Stanford posted a criticism of the choice of UMAP to visualize ancestral diversity.
There was also some excitement in the drug development space. Scientists from Intellia Therapeutics published the results of the Phase-1 trial of their in vivo gene editing program for hereditary angioedema, a once-and-done treatment. The readouts were nothing short of impressive, which you can appreciate from the lived experience of Bev Tanner from Auckland, one of the 10 participants who benefitted from the trial. What looked like science fiction not long ago is now becoming reality—rewriting the genetic destiny to permanently "cure" diseases.
It turns out editing the genetic code is not the only one-shot treatment to silence a gene for lifelong. You can also accomplish the same by editing the epigenome that governs the gene expression programs. A study in Nature last month reported on successful, lifelong in vivo epigenetic silencing of PCSK9 in mice.
We are witnessing the unfolding of the magical powers of gene editing technologies. As a geneticist, I am more excited about the indications—decades to centuries-old genetic conditions—that the companies handpick to showcase their new drug technologies, like how Vertex picked sickle cell disease and beta-thalassemia to successfully develop and deploy an ex vivo gene editing treatment in the clinic, like how Alnylam picked an ultra-rare inborn error of metabolism with a clean liver target to develop the very first successful RNAi treatment for a rare disease, how Intellia picked hereditary angioedema to showcase their in vivo gene editing technology etc. This is what I describe as 'finding problems for solutions', where human genetics and geneticists play an important role in the drug discovery field, a topic that I'd like to deep dive into in a future post. But for now, let’s talk about our regular topics. Below I summarize some of the exciting studies that I read and enjoyed last month organised under various themes as usual. Enjoy
Allelic series
The term “allelic series” was coined in 1963 by Victor A. McKusick, the founder of Mendelian inheritance in man (MIM) (and its online version OMIM), who is known popularly as "the father of medical genetics". It was the time when the medical genetics field just began to move away from the 'one gene one disorder' view with the slowly unfolding realization that seemingly different phenotypes can stem from the same biological defect mapped to a single gene and the phenotypic differences are simply due to the differences in the severity of the mutations. The example that inspired McKusick to write his seminal article on the "Phenotypic diversity of human diseases resulting from allelic series" was mucopolysaccharidoses (MPS). If you take a quick look at the MPS types in Wikipedia, you'll notice that type V is missing. It's because initially it was believed that MPS V is caused by a different gene than the types I to IV, but later it turned out it was caused by defects in the same gene (IDUA) as type I but just a milder form.
Today, we use the term allelic series to describe not only the variants in a gene that produce different phenotypes but also the ones that produce the same phenotype. With the advancements in the techniques of genotype to phenotype mapping, it is becoming clear that the phenomenon of allelic series is ubiquitous and can serve as biological evidence to strengthen the association of a gene with a phenotype. It is something we drug target hunters always look for.
The breakthrough discovery of PCSK9 (refer to my 2022 roundup on the background story) was based on the concept of allelic series. PSCK9's link to cholesterol was first discovered by Catherine Boileau based on the gain of function mutations identified in French families affected with an autosomal dominant form of hypercholesterolemia. Boileau's discoveries then inspired scientists in the US to hypothesize that if the gain of function mutations in PCSK9 increase cholesterol, then loss of function mutations should do the opposite, resulting in the breakthrough discovery of the association of loss of function mutations in PCSK9 with decreased cholesterol and cardiovascular protection. Notably, the discovery was made in a study of a mere 128 individuals of African ancestries. Allelic series happens when there is genetic diversity in a gene, which, as you can guess, can be easily achieved by studying individuals from diverse ancestries. One of the main uses of sequencing individuals from diverse populations is not gene discovery (which is often sample size driven with some rare exceptions) but strengthening the discoveries that were already made and adding insights into the genotype-phenotype relationships, a topic that I’m hoping to write in detail someday.
Below is one of my favourite pictures that I often share on Twitter. The forest plots in the image show the associations of various rare functional variants in a set of genes with relevant traits (e.g. PCSK9 with LDL cholesterol) in the UK Biobank. Each of the variants represented in the figures is driven by a unique set of individuals. This is the level of genetic evidence those who use genetics to do drug discovery aspire for.

Two fascinating papers from very different topics that came out last month reminded me of the concept of allelic series. One was on the topic of evolution and the other was on neurodevelopment. In both cases, researchers discovered new variants in old genes, increasing the resolution of our understanding of the biology of the respective genes.
In the first paper, which is a preprint, researchers from the Broad Institute report on the discovery of noncoding deletion hitting a long noncoding RNA (lncRNA) named CHASERR causing a monogenic neurodevelopmental disorder. The resulting haploinsufficiency of the lncRNA resulted in the over-expression of a nearby neurodevelopmental gene CHD2. It turned out that CHD2 is a dosage-sensitive gene, meaning, both under and over-expression are not tolerated. All previously reported CHD2 NDDs were due to loss of function variants and this is the first time a gain of function is being found. The first author of the paper has written a beautiful thread on the background of the discovery, explaining how they were first intrigued by an innocent bystander mutation, a 5' de novo UTR variant, which led them to the causal mutation upstream: a 22 kb de novo deletion overlapping with CHASERR identified using long-read sequencing.

In the second paper, published in Science Advances, researchers from the University of California report on the association of a coding variant in EPAS1 with high altitude adaptation traits in the Andean highlanders who inhabit the Andes plateau in South America, the world’s most extensive high plateau on earth next to Tibet. As you might have known, EPAS1 is famous in the evolutionary genetics literature for its association with high-altitude adaptation in Tibetans. The variant that was positively selected in Tibetans was a noncoding variant that was introduced from Denisovans to East Asians some 50,000 years ago. It’s an ancient variant and is present in high frequency in Tibetans. Interestingly, the variant that is positively selected among Andean highlanders is a coding variant, which is relatively a lot younger compared to the Tibetan EPAS1 variant, estimated to have been born sometime 10,000 years ago.
The knowledge of the EPAS1 p.His194Arg variant in Andeans itself is not new but the current paper seems to add more to the story by performing functional analysis and adding insights into the origins of the EPAS1 variant. The independent evidence of genetic adaptation to high altitude through EPAS1 variants in humans outside Tibet is an example of convergent evolution where different mutations in the same gene drive the adaptation in different populations. Another well-known example is LCT and lactase persistence where distinct lactase persistence-causing variants underwent positive selection independently in Europe and East Africa.
One other fact that intrigued me is the allelic series in both cases were formed by coding and noncoding variants. As I have mentioned before on Twitter, one of the things that excites me about the field moving from whole exome to whole genome with the emergence of UK Biobank's massive WGS data is the allelic series. There is a good chance that researchers will stumble upon genes where they might be discovering for the first time a gain of function association with a phenotype that has been previously linked with the gene only through loss of function associations or vice versa. This will ultimately advance drug target discoveries.
Long-read sequencing
As I mentioned in my last year roundup, we will be hearing the term long-read sequencing (LRS) a lot this year and in the upcoming years as many clinical and research sequencing projects will start using LRS more often. The literature on the LRS can be grouped in different ways. I usually see them through two lenses.
The first one is my favourite: the clinical genetics lens. Here the researchers use LRS to solve cases that were not solved using the traditional short-read sequencing. This is where the LRS technology was widely used. I have highlighted many examples on Twitter. For instance, in one of my popular posts last year where I summarized a preprint reporting the discovery of the repeat mutation underlying the spinocerebellar ataxia type 4, first identified in a multi-generation family from Utah in 1996. The causal variant and gene evaded detection for nearly 25 years due to the extreme complexity of the disease locus, and finally, researchers solved the case using LRS. It was a GGC triplet repeat mutation within the tenth exon of ZFHX3. One of the studies I highlighted in the 'Allelic series' section—the discovery of non-coding deletion in CHASERR lncRNA causing NDD—is another latest example of genetic discovery made possible by LRS.
The second one is population genetics. Here the researchers use LRS to sequence individuals from the general population at scale to either make genetic discoveries or to create resources that will down the line help researchers to make genetic discoveries. There aren't that many "large-scale" LRS studies so far. In terms of resource generation, the Human Genome Structural Variation Consortium (HGSVC) has been continuing the legacy of the historical 1000 genomes (1KG) project by generating LRS data for a subset (n < 100) of the 1KG samples and expanding the catalogue of structural variations in humans. An example of how this resource fueled downstream genetic discoveries is a paper that I highlighted in my last year's roundup post and also in the year-end episode of The Genetics Podcast by Patrick Short. In that paper, the authors built an imputation reference panel for variable number tandem repeats (VNTR) using the HGSVC LRS reference genomes and imputed VNTRs in the UK Biobank and discovered many disease-relevant VNTRs with impressive effect sizes hiding behind some well-known GWAS loci.
The most impressive population-scale LRS study, however, comes from deCODE Genetics, published in 2021, where the researchers sequenced 3622 Icelandic individuals using LRS and built a reference panel to impute structural variants in their entire cohort (N=160,000). Many fascinating discoveries were born out of this study including my favorite, the association of a VNTR in ACAN with height. The interesting part of the VNTR story, which I have written in this thread, is the genetic signal surfaced a long time ago during the early GWAS studies of height but didn't get much attention despite having the largest effect size until two independent teams (deCODe and Harvard) discovered the extensive repeat variations in the ACAN gene and their impact on the human height.
Speaking of structural variants hiding under GWAS loci, there are two ways such fantastic beasts are typically found.
One is through a hypothesis-free search across the whole genome like how the deCODE Genetics and Po-Ru Loh's team did and uncovered SVs in ACAN (height), TMCO1 (glaucoma), EIF3H (colorectal cancer) etc. The downside of this approach is that the discovered SVs are biased towards the variants that are common and have a large effect size (same story as the early GWAS discoveries) and towards the commonly studied phenotypes.
The other is a hypothesis-based approach where the researchers search for SVs within a specific GWAS locus in an attempt to identify the causal variant. One interesting example is a recent discovery by two independent groups of a structural variant underneath TMEM106B locus, the very first GWAS locus mapped to frontotemporal lobar degeneration (FTLD) in 2010 based on a mere 515 cases and 2500 controls. There were many attempts to solve the causal variant at this locus, including the generation of a knock-in mouse for a TMEM106B missense variant with the belief that this variant is causal and confers protection from FTLD, which however didn't help. And now, using LRS data of TMEM106B locus from healthy individuals, researchers have dug out an Alu insertion variant sitting in the 3' UTR of TMEM106B and is in perfect LD with the FTLD risk variant. Although there is no definite proof that this Alu element is the causal variant, the new finding marks important progress in the long road to solving this GWAS locus.
We discussed about applications of LRS in clinical genetics and population genetics. There is another area where the magic of LRS is starting to unfold: evolutionary genetics. Our genomes are like ancient scrolls, within which are buried evolutionary tales, waiting to be deciphered. Most of the evolutionary insights that researchers have uncovered so far were based on the parts of the genome that were readable using short-read sequencing. With the newly acquired superpower to read parts of the genome that were once invisible such as centromeres, telomeres, the deserts of palindromes in the Y chromosome etc., scientists will be attempting to answer long-standing evolutionary questions. A new preprint on the genetic origins of Down syndrome gives us an idea of what such questions will look like.
In the preprint, the authors sequenced the centromeres of chromosome 21 in an individual with Down syndrome and his both parents in an attempt to answer an evolutionary question: why chromosome 21 is most vulnerable to non-disjunction (failure to separate during meiosis) in humans? The authors found three haplotypes (H1, H2 and H3) in the chromosome 21 centromere sequences of the proband corresponding to the three copies of the chromosome, two of which came from the mother and one from the father. To their surprise, the authors found that the two haplotypes from the mother were strikingly different in length. When studying the centromere characteristics using epigenetic markers, the authors found that the chromosome bearing the longer haplotype had an ill-defined and smaller centromere. So, it's possible that the kinetochore did not attach properly to the longer chromosome during meiosis and as a result, the chromosomes failed to separate resulting in trisomy. It's interesting because, for a long time, it was believed that the small size of the centromere is what made chromosome 21 susceptible to non-disjunction, but the new findings suggest that it's the size asymmetry rather than the absolute size that might be causing non-disjunction. The authors further analyzed the evolutionary age of the identified haplotypes and found that the common ancestor who birthed these haplotypes should have lived not long ago, somewhere around 15,000 years ago. The findings bring more questions than answers and hopefully, will inspire future studies to continue this line of investigation.
Evolutionary History of India
We are in the year 2024. Two decades have passed since the human genome project was completed. The genomic datasets in the UK, Europe and the US have reached unimaginable limits, which have contributed greatly not only to the understanding of the genetics of human diseases and traits but also to tracing human evolution to 50,000 years ago and beyond. While these advances amaze me, they also sadden me when I think about where my country India stands in terms of genomic advances.
Despite housing 18 to 20 per cent of the world's population, India is one of the least represented in human genomic studies both in the context of disease genetics but also in the context of evolutionary genetics. More than 1.5 billion people representing around 5000 genetically, culturally and linguistically distinct communities reside in this vast subcontinent, remaining as a vastly under-explored resource for human genomics. There are many barriers to blame for the slow progress of human genomics in India including political, cultural (ironic that the very characteristic that makes India an invaluable genomic resource is also a barrier to genomic studies), economic and importantly, the mind-setting of some local researchers who are experts in the field who are supposed to be advancing the genomics research by promoting international collaborations but instead spend much of their time scrutinizing and sabotaging foreign collaborations that happen with other Indian researchers.
The good news is the situations are changing, and many exciting progresses are happening. I have highlighted many in the 2022 and 2023 roundups. For example, I discussed a milestone paper based on ~4800 whole genome samples from India, Pakistan and Bangladesh in last year's roundup as well as in the podcast. It's been only 2 months, and we already have an impressive new study that dives deep into whole genome sequences of ~2700 individuals sampled from 18 states across India and traces the evolutionary history of India all the way back to the out-of-Africa migration event. The samples come from a study called Diagnostic Assessment of Dementia for the Longitudinal Aging Study in India (LASI-DAD) established by a team of local and international researchers.
In this work, the authors have performed an extensive analysis of the ancestry, admixture, archaic introgression, natural selection and genetic architecture shaped by ancient cultural practices such as endogamy and consanguinity. The authors bring some clarity to the admixture components and proportions of the two major ancestral groups, the ancestral north Indians (ANI) and ancestral south Indians (ASI) that form the "Indian cline". They also identify outliers of this cline and trace their genetic roots and the migration events coinciding with their arrival in India. The authors identify many novel variants (23 million SNVs and 2.2 million indels) and expand the catalogue of human genetic variations, which will eventually help improve rare disease genetic diagnoses. The authors demonstrate the well-known increased homozygosity in the Indian genomes (particularly in those from south India) driven by endogamy and consanguinity.
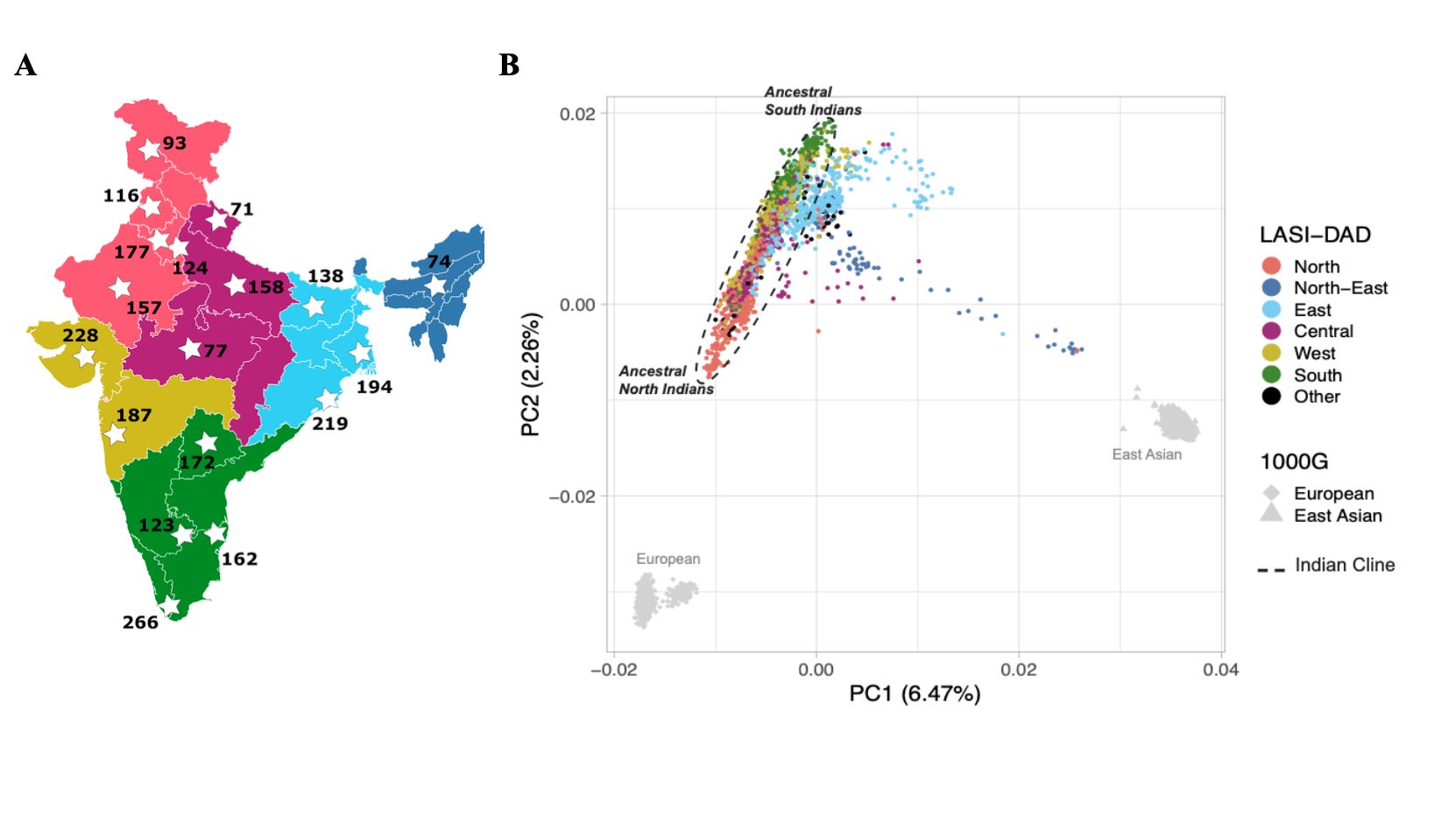
Among all the analyses, I find the work on archaic introgression fascinating. I guess this is the first comprehensive analysis of the contributions of Neanderthal and Denisovan ancestries to present-day Indians. The authors find, of all the world populations studied to date, the Neanderthal segments found in the Indian genomes are the most expansive. Almost all of the bits and pieces of Neanderthal segments that were collected in the past years from the modern human genomes of world populations outside India can be collected from just the Indian genomes. This is also true concerning Denisovan ancestral segments to some extent. The new findings, as per the authors, are surprising and demand a "re-evaluation of models of human origins".
GWAS of type 2 diabetes
I remember the first time I came across a GWAS that surpassed the one million milestone. It was in January 2018 when a bioRxiv preprint on a GWAS of insomnia based on 1.3 million individuals appeared in my Twitter timeline. I was living in Denmark back then. It was the beginning of the final year of my PhD, and I was preparing for my trip to New York for a six-month research stay at Icahn School of Medicine at Mt Sinai. I was so excited by that paper that I spent my entire flight trip from Denmark to New York writing a blog post1 on it. Later I learnt that the first to cross one million milestone was a GWAS of atrial fibrillation that was posted on bioRxiv a few weeks before the Insomnia paper went live.
By now I believe most of us have got past the wow effect that we—at least some of us—used to feel whenever we stumbled upon genetic studies performed on a massive sample size. It is now common for GWASs to report sample sizes in millions. Thanks to UK Biobank, Finngen, Million Veteran Program, 23andMe and recently, All of Us. The largest sample size reported in a GWAS to date was 5.4 million reported in a landmark study of height published two years ago in Nature. I have written about this paper in my 2022 roundup and discussed about it in my 2022 podcast episode with Patrick Short.
As the sample sizes grow massive and massive, it's natural to wonder what's the end game of these efforts. The researchers from the GIANT consortium who published the 5.4 million GWAS of height did answer that question showing that it's possible to saturate a GWAS to the point where all the discovered variants together explain the full common variant heritability of the phenotype. It might be true for a quantitative trait like height, but will never be possible for binary complex disease traits such as type 2 diabetes (T2D), coronary artery disease etc. that often have extreme heterogeneity in their etiologies. So, what is the end game of continuously increasing the sample sizes of GWAS of complex diseases? A massive GWAS of T2D based on 2.5 million individuals published last month attempts to answer that question.
One of the problems of GWAS for complex diseases with widely ranging risk factors (many of which themselves are heritable) is the heterogeneity in the results. That is, the discovered variants not only capture the genes and pathways related to the core biology of the disease being studied but they also capture the genes and pathways related to any heritable risk factors related to the disease. For example, obesity is a major risk factor for T2D2, and obesity in turn has a multitude of risk factors. Lipodystrophy (abnormal fat distribution) is another risk factor for type 2 diabetes. Individuals from certain ancestries, for example, South Asians, store fat around the organs instead of subcutaneous tissue and can easily develop T2D even without becoming visibly obese. Medications that have metabolic adverse effects can cause T2D. And so on. So, a GWAS of T2D will capture genetic signals from all these dozens to perhaps hundreds of different risk factors as a result scrambling our understanding of the biological mechanisms of T2D. This heterogeneity in the results hinders drug development efforts, understanding of the genetics of complications of T2D, genetic predictions of T2D etc.
One way to approach the above problem is to carefully curate the cases and controls by stratifying the individuals based on varied risk factors. While these approaches have been tried in the past, they are not scalable and no matter what, you'll always end up collecting study participants who will be heterogenous concerning one risk factor or the other. An alternative solution to this is what the authors have attempted in the new GWAS of T2D and that is what inspired me to highlight this study.

The authors address the heterogeneity in the GWAS results by focussing not on the phenotype but on the results themselves. By correlating the associations of the hundreds of T2D-associated variants with a range of diseases and traits that are etiologically relevant to T2D, the authors decompose the T2D results into sets of variants based on the characteristics of phenotypes they correlate the most with, for example, T2D variants most associated with obesity, metabolic syndrome, lipid metabolism etc. Now they treat each of these sets of results as if they were derived from a GWAS of individuals in whom T2D was caused primarily by a specific risk factor, for example, the cluster of T2D variants associated with obesity would be like results from a GWAS of T2D caused by obesity and perform downstream analysis such as cell type and tissue enrichment analysis, polygenic score analysis (analogous to pathway-specific polygenic scores) etc. By doing so, the authors have found many interesting findings, for example, they found that polygenic scores based on specific clusters predict certain diabetes complications while the polygenic scores based on T2D in general do not. I found this study interesting not for its results but for its methods, which hint the readers at where the GWAS field is heading in the coming years.
Evolutionary tails tales
We have come to the tail end of this post. I initially thought of cutting this section off. But then changed my mind.
Stories about random genetic mutation events adding new branches to the evolutionary tree will never cease to blow scientific minds. A recent paper in Nature on how humans lost their tails has been making headlines everywhere last month. A curiosity-driven investigation has led the scientists from NYU School of Medicine to the discovery of a jumping gene (an Alu transposable element) that might have birthed our tailless ancestors from whom we evolved. There are hundreds of news articles on this story. So, I don't feel the urge to go into details. The best places to learn the background story of this research are the Twitter threads and podcast episodes from the last author Itai Yanai, a celebrity scientist on Twitter known for his 'Night Science Podcast' and the first author Bo Xia who conceived the research idea after being struck by inspiration one day while sitting on the pain from his tail bone injury. However, I feel compelled to highlight the beauty of the genetic design underlying the discovery.
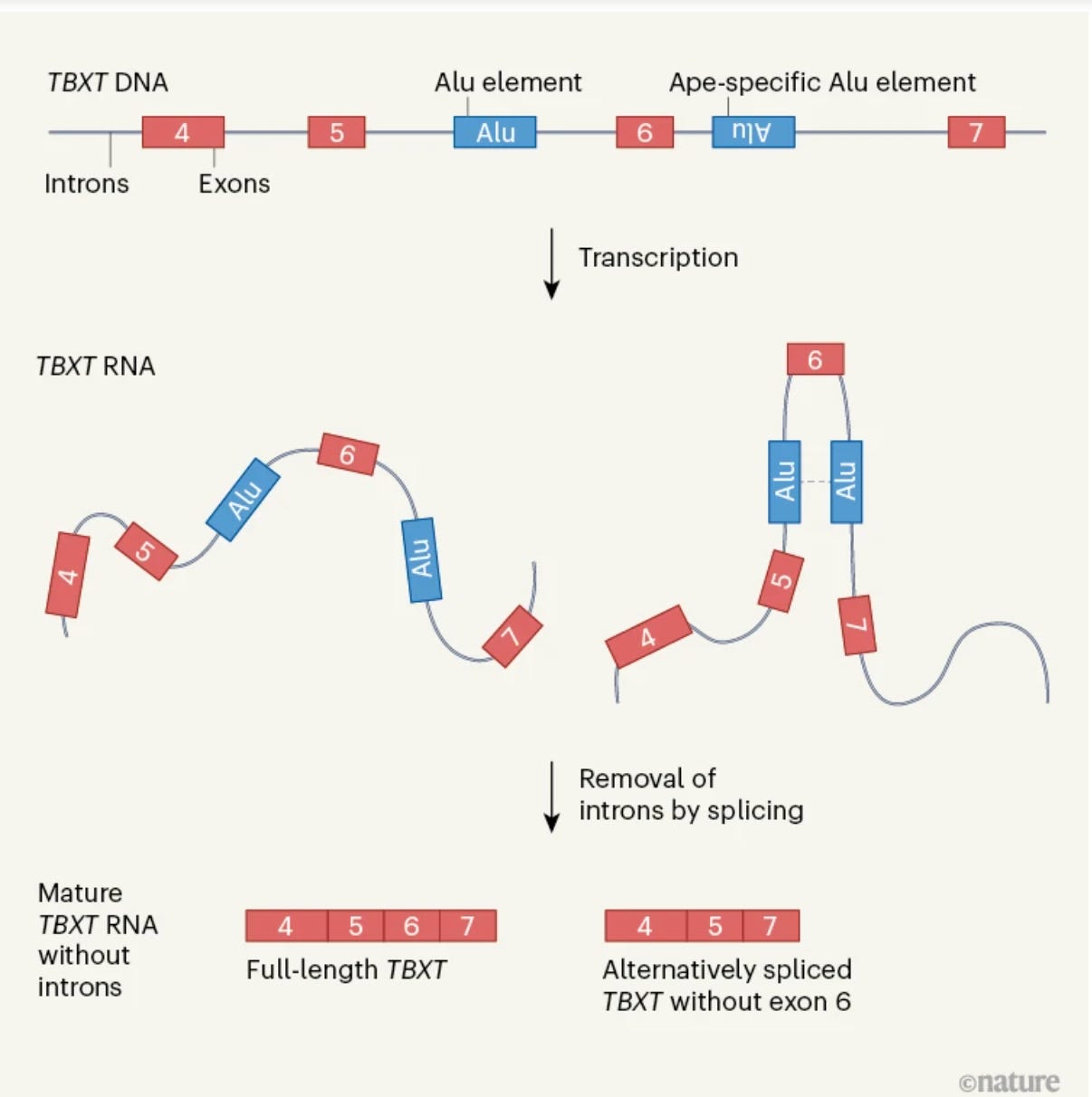
Some of the most elegant designs to tinker around the genes can be found in Nature. Discoveries like the one above allow us to pause and savour the beauty of evolutionary designs—perfections born out of accidents. The mutation event the authors have uncovered was an insertion of an Alu sequence within an intron of a transcription factor gene TBXT. I guess the awe moment was not when the authors stumbled upon the Alu sequence, but when they realized that it wasn't there alone. There was another one in reverse orientation (not hominoid-specific), a partner in crime, hiding within the adjacent intron. The magic happens only when the two partners join hands after the gene is transcribed. After the nascent mRNA is formed, the two Alu sequences bind to each other due to their reverse complementarity and trap the in-between exon 6 in a loop, ultimately excising it away from the mature transcript. The loss of this exon 6 is what appears to be the critical event that resulted in the tail loss.
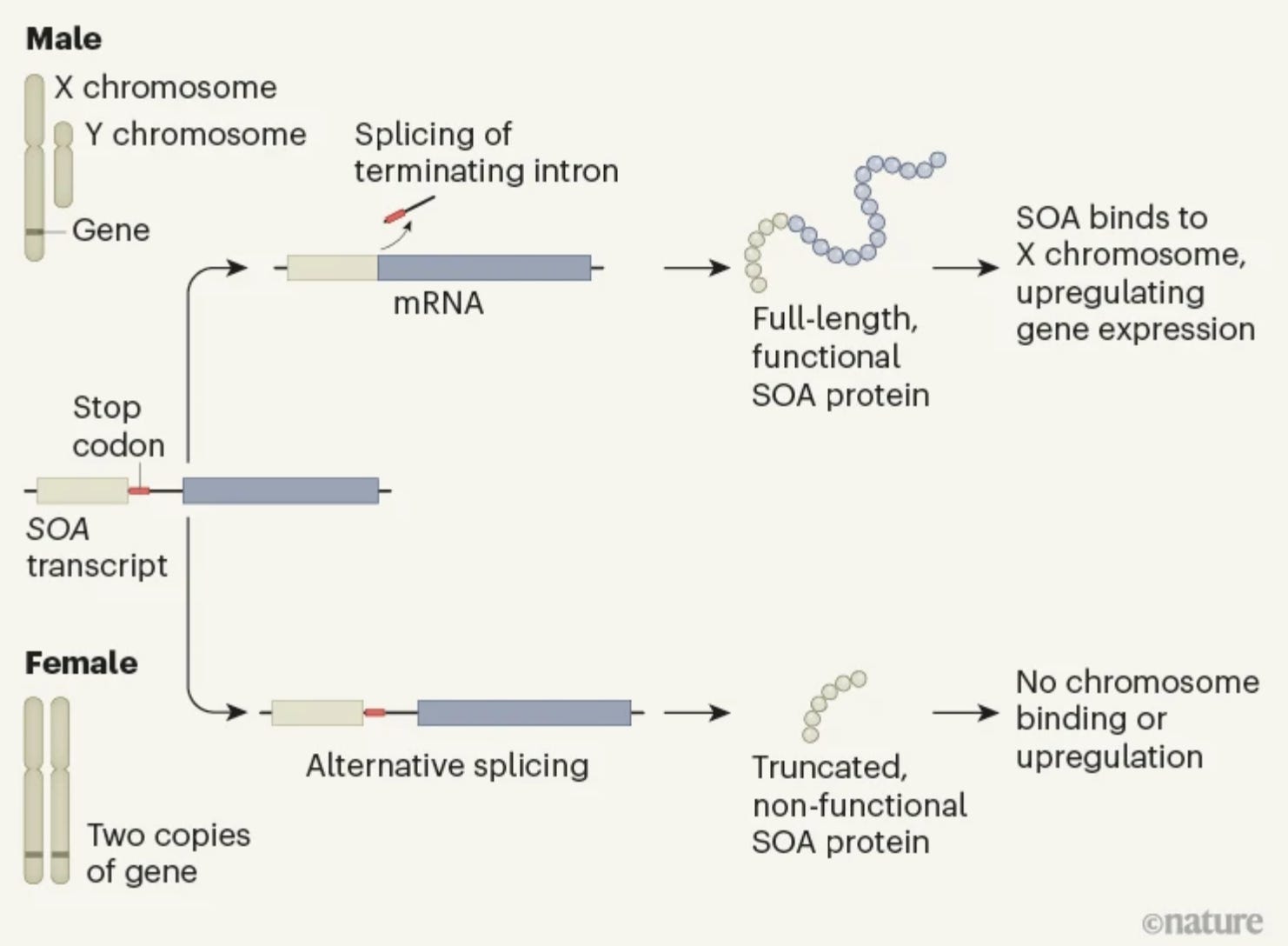
Reading about the exon skipping design reminded me of a similar evolutionary design reported in Nature last year by scientists from the Institute of Molecular Biology, Mainz, Germany. In their work, the authors discovered the master regulator of X chromosomal compensation in anopheles mosquito. Unlike in humans where the female compensates for the extra X chromosome by randomly inactivating one X copy, in mosquitoes, males do the heavy lifting. They double the expression of all their X chromosomal genes to match with the females, and this process is under the control of just two genes--Yob and SOA. The star of the show is SOA (sex chromosome activation) whose expression is 100-fold higher in males than females during embryo development. This extraordinary difference in the expression between sexes is accomplished through alternative splicing caused by the retention of an intron in the mature transcripts in females but not in males. It turned out there is a stop codon in the intron sequence and as a result, the mRNA translation stops prematurely in females resulting in a truncated protein while the male embryo produces full-length functional protein kickstarting the dosage compensation.
And that’s a wrap. I hope you enjoyed the post and learnt something new. More exciting stories to come in the next roundup. Stay tuned! Thanks for reading GWAS stories :)
—Veera

I deleted this post, which was published in Medium, a couple of years ago while deleting my account.