
In pursuit of recessive genes
How Abraham Wald's work inspired human geneticists to find recessive genes

Happy Friday! I hope that at least some of you have read my January human genetics roundup. For this week’s From the Twitter archives post, I chose a Twitter thread that is closely related to one of the papers from deCODE genetics I highlighted in the roundup. As I mentioned in the post, the deCODE scientists have special interests in finding recessive genes (which is not surprising as, you know, Iceland is a founder population). The deCODE team has published a series of papers on this theme and today’s post highlights one of them that was published in Feb 2022. It was the time when “Twitter” was still Twitter and I was writing real threads, which you can appreciate from my liberal use of screenshots from the paper.
I love this paper a lot for two reasons: first, the obvious one, the beauty of the study design and the depth of the investigation, and second, the thread I wrote had a surprising impact, which I discovered a few months later after I posted this thread. On May 18th, deCODE Genetics organized a two-day conference for its 25th anniversary (do you know who was the host? Magdalena Skipper, the Chief Editor of Nature!). They posted the videos a few days later, and when I was listening to the talks, I was pleasantly surprised to see the Punnet square image that I made for the Twitter post was displayed in the slide presented by the senior author of the paper, Patrick Sulem, the head of clinical sequencing at deCODE, whom I admire and respect greatly. Later I heard from Patrick that he and others read my thread and enjoyed it, and he particularly liked the idea of using the Punnet square and so, used it in the presentation. It was one of my proud Twitter accomplishments.
From the Twitter archives

I've been wanting to thread this paper since the day I saw this post. Writing about two great papers on mutational constraint this week has finally inspired me to sit and write about this fascinating work from deCODE. The current paper is one of the many that elegantly show why deCODE is one of the best places in the world to do human genetics.

Having genetic data for >40% of Icelanders linked to extensive medical and population registries along with near-complete genealogical information of the whole country for the last century makes deCODE truly "a global leader in human genetics".
I am sorry to share this picture again. But I find it impossible not to share when talking about mutational constraint, particularly when talking about the current paper.

Abraham Wald's work during World War II has revolutionized many fields, including human genetics, as beautifully explained in this paper on constrained coding regions from 2018.

Wald predicted that the parts of the plane that are least damaged are the most critical. But, imagine, if Wald could go back into the war field and examine the debris of the shot-down planes and confirm his predictions. That is exactly what this paper is about.
In the current paper, the authors focus on a specific set of genes that are depleted of homozygous mutations but not of heterozygous. Such genes typically cause recessive genetic conditions.
If you've attended genetics courses you might remember the Punnet square exercise where you calculate the probabilities of offspring genotypes given the parents' genotypes. Here is an example showing the genotype probabilities of a child born to parents who both carry one copy of an allele (A) that causes a severe recessive condition.
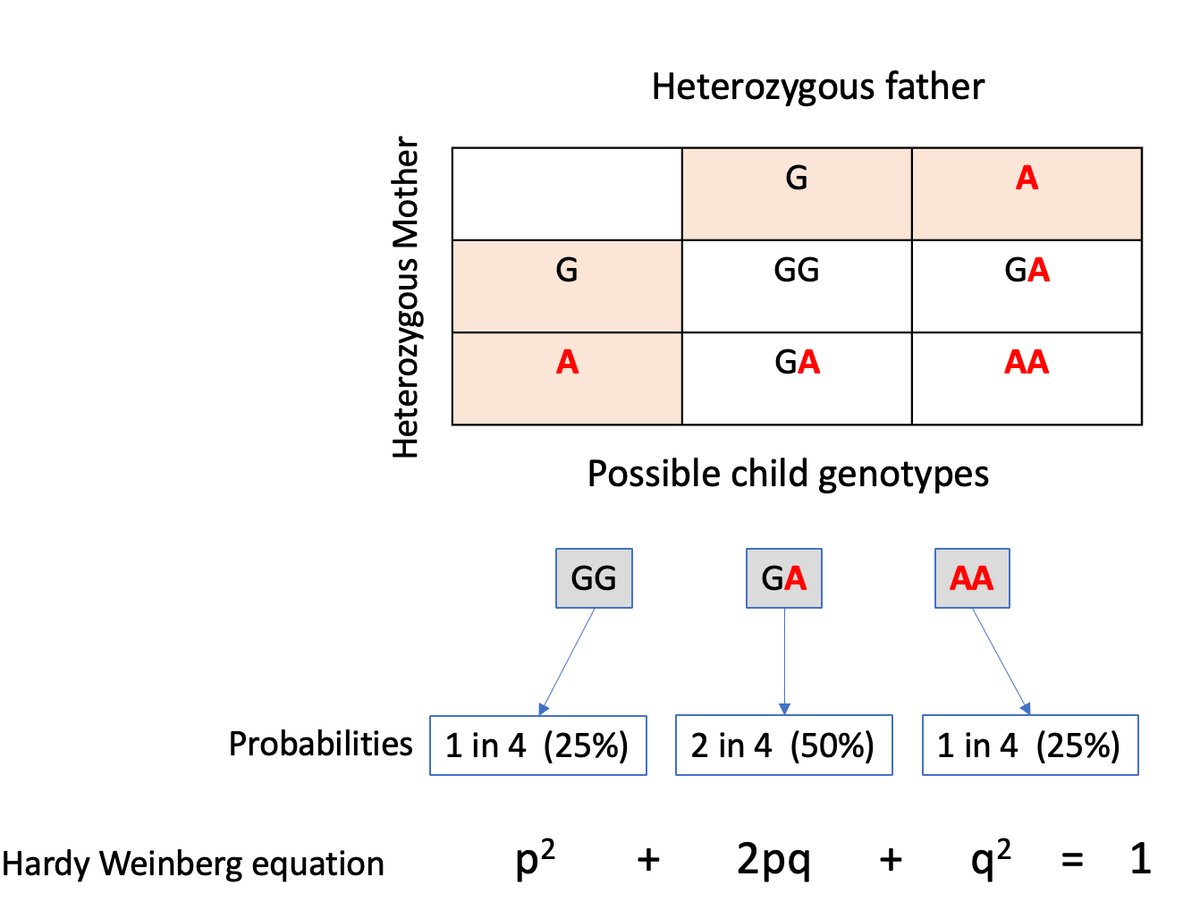
If you take all such carrier couples in a population, under Hardy-Weinberg equilibrium, you'd expect 25% of their offspring to have the genotype AA. But as this genotype is lethal, in reality, you'll see extremely few or no AA homozygotes, suggesting a deviation from HWE.
Identifying genes that are depleted of homozygous loss of function variants (pLOF) in a population (relative to heterozygotes) is one way to discover recessive genes, which deCODE has demonstrated previously in their famous human knockout paper. However, doing the same for missense variants is challenging as their effects on proteins are not as clear as often as it is for pLOFs. In the current paper, the authors take up this challenge to recessive disease-causing missense variants.
Scanning through genotypes of ~150,000 Icelanders (adults, mean age 58.5), the authors were able to find 114 missense variants with high MAF ( >40%) but with zero homozygotes (under HWE, there should be at least 3). 34 of these missense variants are in genes known to cause Mendelian recessive diseases (cystic fibrosis, glycogen storage diseases, encephalopathies, nephrotic syndrome etc.)

Interestingly, homozygous carriers for five of the 114 missense variants were seen in a separate clinical cohort of 764 individuals who underwent diagnostic whole genome sequencing, which gave the authors a unique opportunity to map these genotypes to their phenotypes.

As you can see from the above table, four of these genes already have a known recessive disease reported in OMIM. The remaining one, CPSF3, which has no disease conditions mapped so far, is the star of the show.
In the clinical cohort, two children (A and B) were homozygotes for CPSF3 and had strikingly similar clinical features--intellectual disability, developmental delay, seizures, and nystagmus. A genetic diagnosis was not established for both previously.

This is where things get more interesting. Inspired by the above finding, the authors went back to their original genetic dataset of ~150k Icelanders and identified 3 couples who were carriers of the CPSF3 Gly468Glu variant. Using their genealogical database, they were able to identify all the offspring who were born to these couples. If we go back to our Punnet square again, there is a 1/4 chance for any of the offspring born to these couples to be homozygous for Gly468Glu.

All combined there were 10 offspring, 4 of whom (C, D, E and F) died before 8 years of age. All 4 offspring's clinical features were fascinatingly similar to patients A and B's clinical features. Wait, this gets even more interesting.

The authors were able to get paraffin-embedded archival samples for two of the offspring--C, born 1960, died 1967 and D, born 1964, died 1968--and confirm the homozygous state of Gly468Glu using WGS and Sanger sequencing.

Well, the story doesn't end there. Through GeneMatcher, the authors identified two more patients in the US who are homozygous for another missense variant in CPSF3 but with the same clinical presentation--ID, microcephaly, and seizures.

Altogether the authors were able to establish genetic diagnosis for 8 patients--three of them born in a different era (1950s and 60s) and two of them born in a different continent. That's truly amazing.
So what gene CPSF3 do? CPSF3 codes for a subunit of the Cleavage and Polyadenylation Specificity Factor (CPSF) complex, which is essential for mRNA preprocessing (cleavage and polyadenylation) before they are exported out of the nucleus.

The Discovery of this CPSF3 missense variant marks the first Mendelian disease linked to the CPSF complex. Now we know what happens if the mRNA export from the nucleus to the cytoplasm is disrupted.
Interestingly, someone had already discovered an anti-leukemia drug that acts by inhibiting the CPSF3 complex without even realizing its mechanism of action (was realized only later).
It turned out that the drug inhibits CPSF3 and blocks the mRNA export thereby leading to cancer cell apoptosis. I love how the work of scientists from different fields converges on the same biology.
There are two other fascinating genetic investigations reported in this paper, but I'll stop here. This is a truly remarkable genetic paper and easily one of my favourites.
