
Time travel inside Huntington's brain
A cell by cell exploration of Huntington's disease progression


Happy Friday! One of the talks at the ASHG 2023 blew the minds of the audience. It was a talk on single cell sequencing of brain tissue from Huntington's patients, presented by Bob Handsaker from Steve McCarroll's group at the Broad Institute in Boston. I didn't grab the whole story during the presentation. But the parts that I heard and the final revised disease model that Bob presented felt groundbreaking, and I couldn't contain my excitement. I tweeted about it that night, and many agreed with what I felt. I spoke about it with Patrick Short (who was as excited as I was about this work) in the 2023 year-end episode of The Genetics Podcast. However, I feared that I may have overblown the impact of the work prematurely, and the balloon would burst someday when the actual preprint comes out. Like many others, I've been waiting for the preprint to drop.
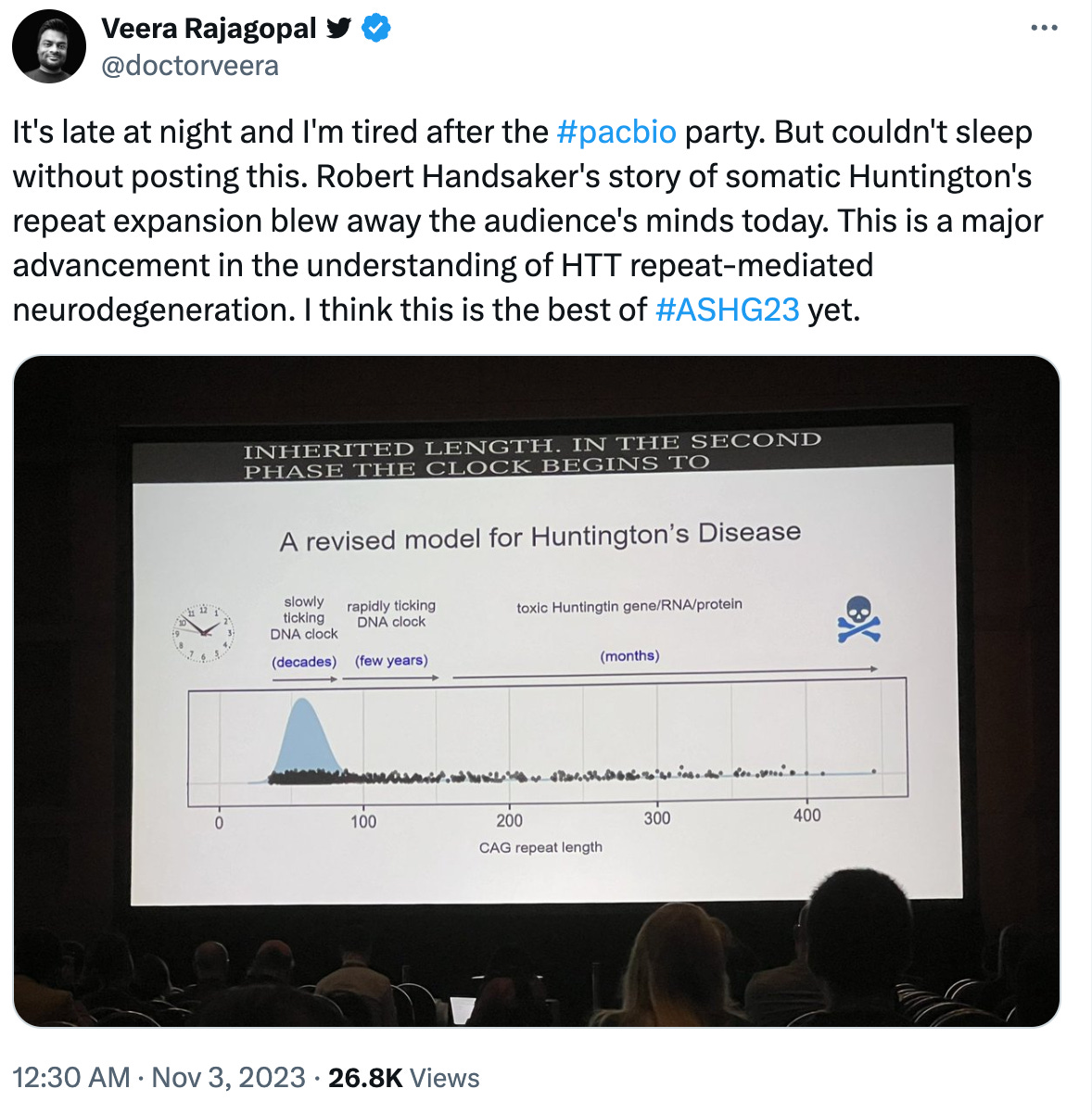
Steve's team finally posted their work in the medRxiv a week ago. Having now read the paper fully, I can say with full confidence that the hype that Bob's ASHG presentation received is well deserved. The work described in the preprint is as just as impressive as I imagined. The work has already garnered a lot of excitement in the community. Here is a tweet from Mark Daly, a reputed, world famous scientist in human genetics. I don’t remember hearing such a praise from Mark on any work before.
What’s known?
It's important we first understand what's already known to appreciate what's new. Huntington's disease is caused by expansion of a microsatellite mutation--CAG repeat--located in the exon 1 of HTT gene in chromosome 4. The number of CAG repeats in the general population range between 15-30, whereas in Huntington's patients it is between 36 and 55.
The CAG repeats in the HTT gene transcribe and translate into poly-glutamine chain in the Huntingtin protein; the HTT is expressed in the fetal and adult brain, among other tissues. The huntingtin protein with an elongated poly-glutamine tract is neurotoxic. It erodes the neurons, particularly the neurons of the striatum, over time in the Huntington's patients, resulting in motor, cognitive and psychiatric symptoms.
Huntington's is a dominant disease. Elongation of only one allele is sufficient to fry the brain. A second hit doesn't have any addition impact on the disease course. There is a long latency period. The age of onset is around late adulthood, typically around 40 years. It takes decades before the symptoms manifest. Though we don't know what causes this latency, we know that the age of onset correlates with the CAG repeat length; longer the repeats, earlier the disease onset.
The CAG repeats in HTT expand across generations (in the germline) but also within a generation (in somatic cells). The phenomenon of expansion across generations is described as "anticipation", where the disease becomes severe in subsequent generations. The germline expansion occurs as a consequence of DNA polymerase slippage, resulting in strand loops. The DNA mismatch repair proteins while attempting to fix the loop end up adding more DNA base pairs than what was originally present, resulting in repeat expansion.
The somatic expansion mechanism is less understood than the germline expansion, particularly in the post-mitotic cells such as neurons. One theory is the expansion is a consequence of limitations of the DNA repair system to properly fix the DNA strand loops that happen during gene transcriptions, particularly genes with repetitive regions like HTT. Instead of excising the lengthy strand and synthesizing a matching short strand, the confused DNA mismatch repair proteins do the opposite: excise the short strand and synthesize a matching long strand. This theory is strengthened by the fact that genetic variations in mismatch repair genes modify the penetrance of HTT repeats.

What’s not known?
Of all the brain regions, why striatal neurons are specifically affected in Huntington's disease?
Clinically, a threshold of 36 repeats in the germline is considered pathogenic. But at the molecular level, what is the threshold for pathogenicity?
It's known that somatic repeat expansions are contributing to neurodegeneration. But how? Are somatic expansions the primary mediators of neurodegeneration, or they simply modify the severity and rate of neurodegeneration?
Why is there a long latency? Is it because the toxic huntingtin proteins slowly kill the neurons, or they wait for a period of time (perhaps to become long enough to hurt) before starting to devour the neurons?
At what time point, the neurodegeneration becomes completely irreversible? Is there a therapeutic window of opportunity to reverse or halt the disease progression?
The current paper
The current work by Handsaker et al. doesn't answer all the unknowns. But they add major insights into the missing pieces of the Huntington's puzzle through single cell sequencing of brain tissue from ~60 Huntington's patients and ~50 controls. I was never a big fan of single cell sequencing technology, as I always felt that it was hyped more than it delivered. This is probably the first time, I was able to truly appreciate the value of this technological advancement.
In a nutshell, by studying the mRNA transcripts of individual brain cells of patients with varying degrees of neurodegeneration, the authors were able to model the molecular disease course of Huntington's disease.
Loss of striatal neurons
Firstly, the authors demonstrate the well known selective death of striatal projection neurons (SPN) in the brains of Huntington's patients. Look at the dramatic reduction of SPN proportion in the caudate nucleus (most affected part of the striatum) of the Huntington's patients in below plot.

Inherited CAG repeats vs SPN loss
Next, the authors demonstrate the relationship of SPN loss with age and CAG repeat length. They use a standard metric called CAG-age-product (CAG) score that reflects the cumulative exposure of a Huntington's patient to expanded CAG repeat. It is calculated by multiplying age with germline CAG repeat length minus 33.66. The relationship between CAP score and SPN loss in the below plot captures both the latent period (CAP< 300) when the SPN loss is only moderate and disease onset (CAP> 400) when there is rapid neurodegeneration. At advanced stages (CAP>600), most of the SPNs (>80%) are lost.

Somatic repeat expansions
Having established the known relationship between SPN loss and germline CAG repeats, the authors turn their attention to somatic repeats. The authors quantify the CAG repeat length directly from the HTT transcript in each of the brain cells. The authors found extensive somatic expansion of HTT CAG repeats in the SPNs, but only mild expansions in the other cell types. This is an important finding, and it suggests that the selective death of striatal neurons is not because they are more vulnerable to polyglutamine toxicity than other cell types, but because huntingtin protein with extremely long polyglutamine tract is produced only in the striatal neurons. Should such toxic huntingtin proteins are produced in other cell types, they also will likely die, which is what we see in Huntington's mice models with extra-long CAG repeats in the germline.
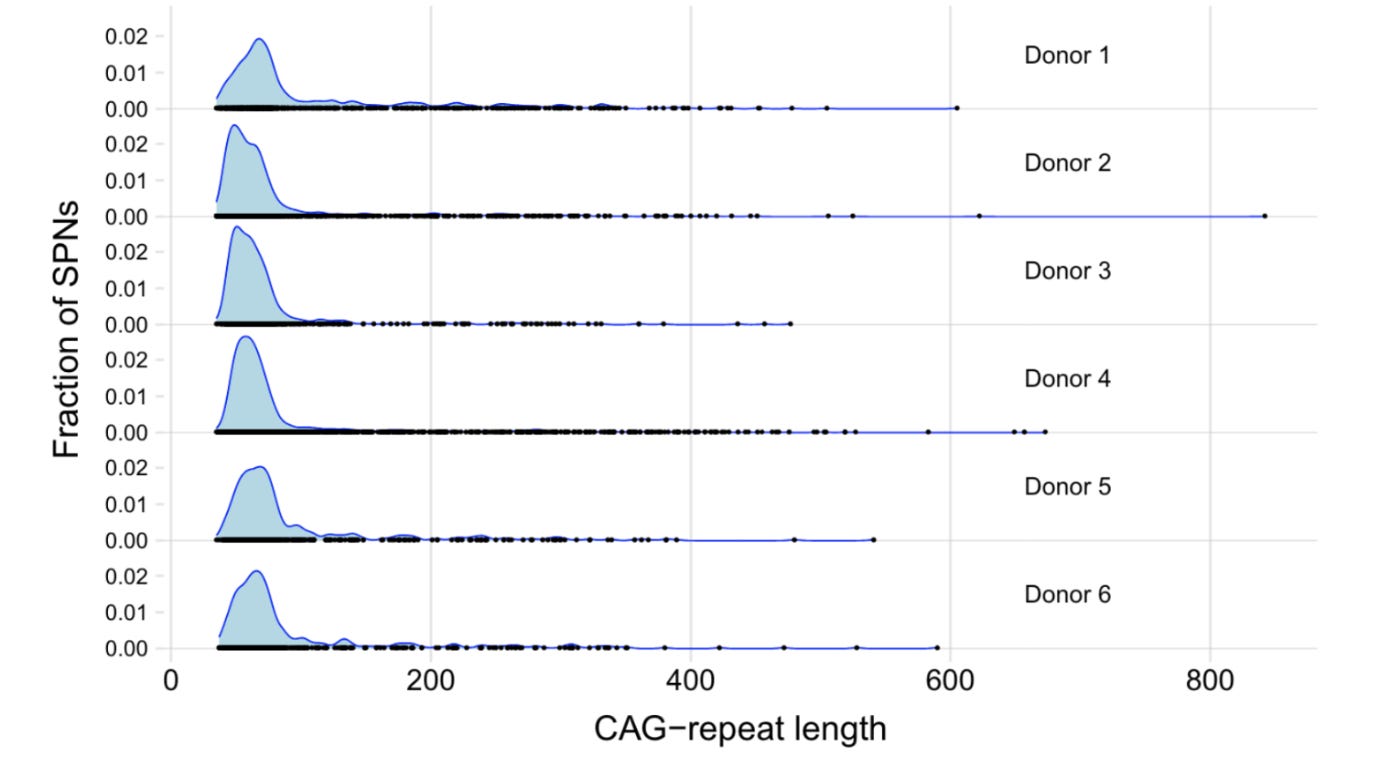
Armadillo distribution
The authors found that not all the SPNs showed similar expansion. While the bulk of the SPNs showed a moderate expansion (around 20–30 repeats more than the germline), a tiny proportion of the SPNs showed extreme expansions (around 100-500 more than the germline). The two groups form a characteristic distribution shape, which the authors compare to an armadillo. They call the body of the armadillo, phase A and the tail, phase B. It seems earlier studies failed to capture the extremely elongated CAG repeats because of technical limitations of the PCR methods.
Repeat expansion vs gene expression
This is the most exciting (and innovative) part of the work. The authors compare gene expression changes between different repeat lengths. Thanks to somatic mutations, each person has their own allelic series of diverse CAG repeat lengths. The authors leverage this fact to perform a within-person comparison of gene expression changes (as a read out of pathogenicity) across different CAG repeat lengths and avoid all the confounders that will arise when comparing between persons. That's a brilliant idea! The results of this analysis are the core findings of the paper.
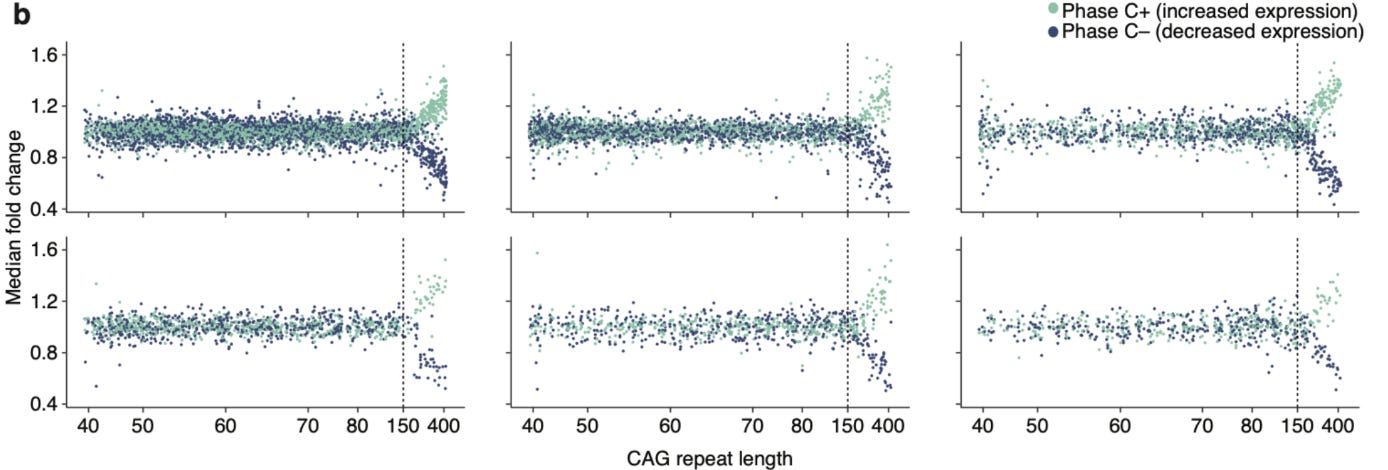
The authors find repeat expansions of up to 150 repeats had no major impact on the gene expressions. But beyond 150 repeats, there is a dramatic impact, distorting the expressions of hundreds of genes.

Wait, here is the most interesting part. They found that the gene expression changes are highly reproducible across individuals. Look at this correlation plots. Incredible!

Next, the authors trace how the gene expression distortion worsens with increasing CAG repeats. Based on their observations, they define different phases. Remember, we have already seen phase A (the body of armadillo with <100 repeats) and phase B (the tail of armadillo with >100 repeats). The authors now add more phases to the tail.
The phase B ends at 150 repeats, the magic number below which there is no much harm, but once past 150, things go south quickly. The authors bucket the neurons with repeats >150 in three phases: C (Continuous escalation), D (De-repression) and E (Elimination).
During phase C, with expanding repeats, there are continuous changes in gene expressions; they get more and more chaotic to a point where neurons lose their cellular identity. The gene expression patterns of individual cell types are like fingerprints. If you're familiar with scRNA-seq paper, you'd remember seeing tSNE plots showing clusters corresponding to different cell types. Such identities are lost in phase C. At this point, you can no longer tell apart striatal neurons from other neuronal types, or even from a non-neuronal cell type.
Next comes the phase D, the de-repression phase. Now, zombies begin to crawl out of their graves. Genes that are normally disallowed in the striatal neurons start expressing because of the loss of repression. At this point, the CAG repeats have surpassed 350 and the clock is ticking much faster. The authors explored what sort of genes are de-repressed. They found they are mainly transcription factors and noncoding RNAs normally expressed during early embryonic development but not in the adult neurons. It’s as if the neurons are aging in reverse all the way to how they were inside the womb before disappearing forever.
Then comes the final phase, phase E, the elimination phase1. The neurons now have reached the end of their life and so does the patient. Almost all the SPNs are lost and the caudate nucleus is fully atrophied, ultimately resulting in the patient's death.
ELongATE neuropathology model
Putting all five phases together, the authors propose a new model of neuropathology of Huntington’s disease called “ELongATE” (extra-long repeats acquire toxic effect) where the striatal neurons go through five phases before their death.

The slowly, capriciously ticking DNA clock.
During the phase A, the HTT CAG repeats expand slowly and aynchronously. The authors write “We estimate that an SPN takes 50 years (on average) to expand from 40 to 60 CAGs, then another 12 years to expand from 60 to 80, …”.
“Asynchronous expansion” is the key term. The repeat expansion is a stochastic process. So, rate of expansion of one neuron is completely random compared to an another. Yet they all expand slowly and spend >98% of their lifetime in the phase A (armadillo body). The authors compare phase A to “a slowly and capriciously ticking DNA clock.”
The rapidly, predictably ticking DNA clock.
When the neurons reach phase B (80 to 150 repeats), their repeat progression become predictable. As the CAG repeats become longer, their chances of undergoing new expansion mutations increase. As the neurons step inside phase B their life course becomes predictable, and their pending years of life can be timed. The authors compare phase B to “a rapidly, predictably ticking DNA clock”.
Interestingly, even during the rapidly progressing phase B the neurons still seem to function and so, there are no symptoms. Only after entering phase C, the neurons start eroding (as indicated by the gene expression distortion), resulting in symptoms. And quickly they pass through phases D and E.
Therapeutic window
Based on their analysis, the authors predict that there is a long therapeutic window of opportunity during which any interventions that can halt the repeat expansion can be effectively made to delay the disease. The authors discuss that even after the symptoms onset, the disease progression could be slowed down by rescuing the bulk of the phase A neurons from entering the next phase.
Stopping repeat expansion is the key
Based on the findings, it looks like the therapeutics for Huntington’s should be focussed on stopping the repeat expansion. Past drug development efforts focussed on reducing the toxic huntingtin protein, which didn’t work. Both human genetics and animal models have shown that interfering with mismatch repair system slows down the disease. Likely, many companies are currently working on targeting mismatch repair genes to treat Huntington’s disease. If such therapeutic designs turn out safe and effective, we will be seeing a newer generation of miracle drugs for not just Huntington’s but many other repeat expansion-related neurodegenerative disorders.

The elimination phase E is extrapolated based on the observed data, as you cannot study neurons that are dead.