
Discovery of TMEFF1, a viral restriction factor in the human brain
Exome sequencing of individuals with herpes simplex viral encephalitis identifies a new genetic mechanism through which neurons resist viral infections

I ended my previous story with the take home message that the drug discovery field hasn't explored the within-individual biological diversity as much as it has explored the between-individual genetic diversity. And this week, we have another remarkable story that further emphasizes my point.
After completing MD in 2014, I moved from India to Denmark to do a PhD in psychiatric genetics. My interest in psychiatric genetics came from working on a related topic for my MD thesis. That was my entry into the world of human genetics. At that time, I knew little about the field. During my PhD, I learned about the genetics of ADHD, autism, schizophrenia, and other psychiatric diseases. Later, when I began reading broadly, I felt a longing towards certain areas of human genetics like cardiovascular system, endocrinology and metabolism. As I read more, the list grew longer. The latest addition to the list is infectious diseases, particularly the ones that affect children.
Ever since I discovered Jean-Laurent Casanova, an amazing physician scientist at The Rockefeller University, I've been on and off reading about monogenic infectious diseases. Casanova has made tremendous contributions in this area. The first time I learned about him was in early 2022, when I came across two parallel publications in The Journal of Experimental Medicine on the discovery of null mutations in interferon genes that explained the high vaccination-associated deaths in Polynesian Islands and circumpolar regions. I've written about this story previously.

Human immune system has been evolving for over hundreds of thousands of years through continuous arms race against viruses, bacteria, fungi and parasites. For a pathogen, for example, a virus, to infect humans, it has to bypass multiple layers of security measures. It needs to first enter the body, then the cells, then the nucleus if it requires access to host's transcriptional machinery. There are sophisticated sensors in both inside and outside cells that monitor for foreign intruders. Upon detection, they unleash an army of proteins that attack the virus from different angles and chop them into pieces. How scientists first came to know about the genes and pathways that make our extraordinary viral immune defense systems?
I am tempted to say through human genetics. But at least in the case of immunology and infectious diseases that is not entirely true. Even before the reports of gene mutations increasing susceptibility to infections were published, scientists knew a lot about the pathways involved in pathogen immune response through molecular and animal studies. Many of the discoveries of monogenic causes of infectious diseases were in fact driven by prior knowledge of the genes and pathways associated with such diseases. I have highlighted few examples of how mouse genetics played a major role in the discoveries of monogenic causes of systemic lupus erythematosus (SLE).

Autoimmune diseases like SLE represent one end of the double-edged sword that is almost every immune gene. The other end is the immune deficiency. There are countless examples of gene defects that increase susceptibility to viral, bacterial, fungal and parasitic infections. Many of those discoveries were driven by prior mouse genetic findings.
For example, the first reports of genetic defects increasing susceptibility to mycobacterial infection in humans surfaced in 1996. Two parallel studies in NEJM (one of which was from Casanova's group) reported mutations in IFNARG1, encoding interferon gamma receptor 1, abolished immune response to mycobacterial infection, even the ones by attenuated strains. As a result, carriers developed disseminated infections after Bacillus Calmette–Guérin (BCG) vaccination. But even before these reports, scientists knew that deleting any of the genes in the type 2 interferon signaling axis—interferon gamma (IFNG), its receptor (IFNARG1) or its downstream transcription factor protein interferon gamma regulatory factor (IRF1)—makes the mice susceptible to BCG infection. And this knowledge is what motivated Casanova and his team to search for mutations specifically in these genes when studying a child who died of disseminated BCG infection after vaccination.
We examined the five genes coding for interferon-γ, interferon-γR1, IRF1, TNF-α, and TNF-αR1 in a child with fatal idiopathic disseminated BCG infection. We found a mutation of the gene for interferon-γR1. There was no detectable interferon-γR1 on the cells from the affected child. These findings provide further evidence of the importance of interferon-γ in the response to mycobacterial infection.
While human genetic findings in certain scenarios simply confirmed what was previously found through mouse knockout studies, in other scenarios, they brought critical insights that greatly propelled forward the understanding of immune pathways. STAT1 (signal transducer and activator of transcription factor 1) is an important immune protein and is at the cross roads of multiple interferon signaling pathways. Early STAT1 mouse knockout studies focussed mostly on the viral diseases. In 2001, Casanova's group reported the first STAT1 genetic mutation in humans, which increased the susceptibility to mycobacterial infections, but surprisingly, did not increase the risk for viral infections. The two patients with history of disseminated BCG infection were found to have partial STAT1 deficiency due to heterozygous mutations. Just two years later, Casanova's group reported the consequences of complete STAT1 deficiency in humans. Searching for genetic diagnoses in two infants who died of unexplained viral illnesses, the authors discovered that both patients were knockouts for STAT1. The two studies together painted a clear picture of the recessive role of STAT1 in type 1 interferon signaling (induced by alpha and beta), and dominant role in type 2 (induced by gamma).
Our description of infants genetically deprived of STAT-1-mediated responses to IFN-α/β and with lethal viral disease provides the first evidence, to our knowledge, that endogenous IFN-α/β exerts potent antiviral effects in vivo in humans. More generally, our study of humans shows that IFN-α/β is crucial for protective immunity to viruses in natural conditions of infection.
The search for genetic causes of unexplained infectious diseases has always been guided by prior knowledge of the genes and pathways related to the particular pathogen. And in those searches, often scientists found what they expected to find. But sometimes, human genetics can surprise scientists by revealing something completely new, something extraordinary, opening the door to a new therapeutic mechanism.
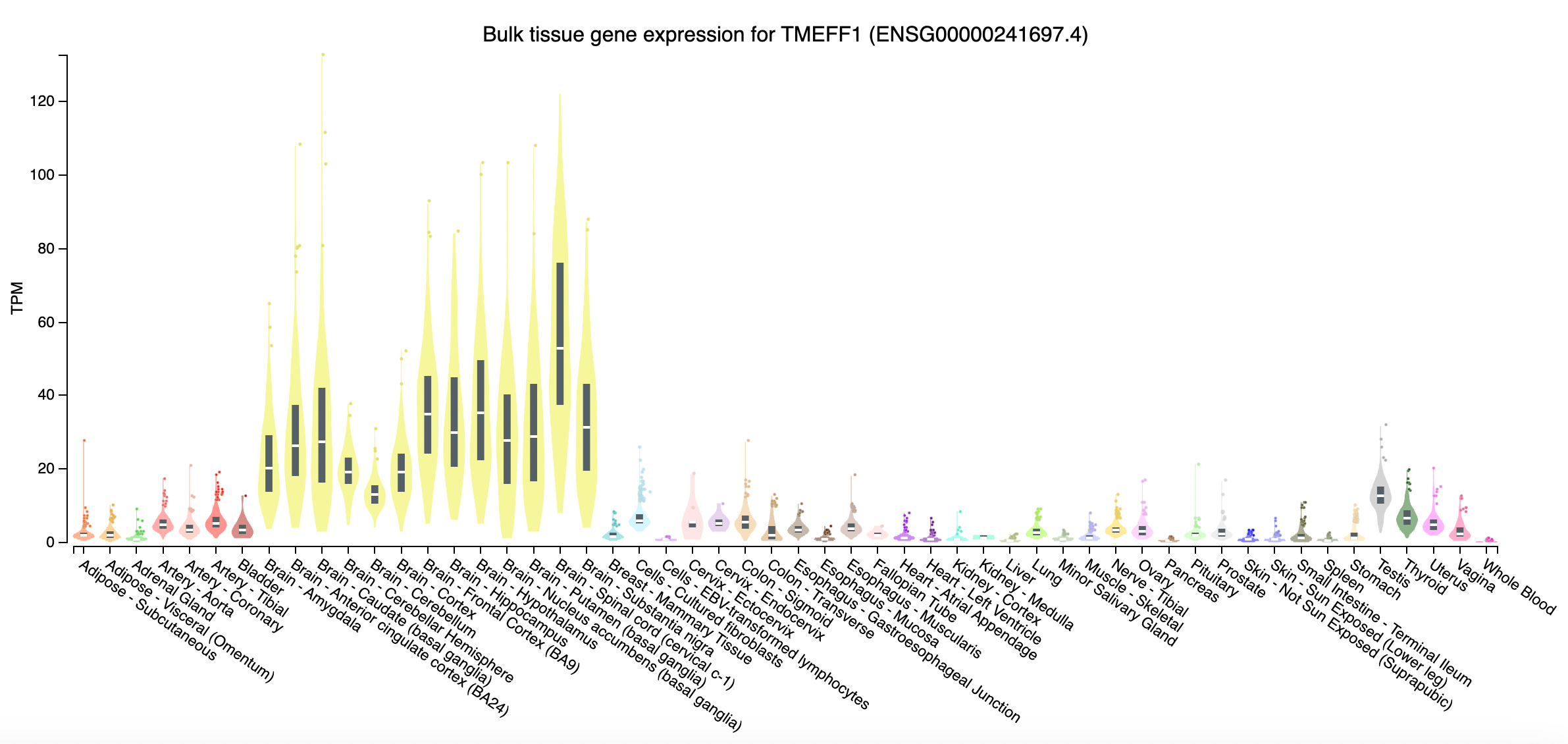
In a new paper in Nature, an international team of researchers led by Casanova and Yi-Hao Chan at the Rockefeller University report the discovery of a neuronal membrane protein, tomoregulin-1, that protects the human cortical neurons from herpes simplex viral infection. The gene TMEFF1 encoding tomoregulin-1 was cloned 28 years ago. Until this year, scientists had little idea what this protein does, except that it is specifically expressed in the brain. Now, studying two unfortunate humans who were born without a functional copy of TMEFF1, scientists have discovered that the gene encodes a viral restriction membrane protein that prevents viruses from entering brain cells.
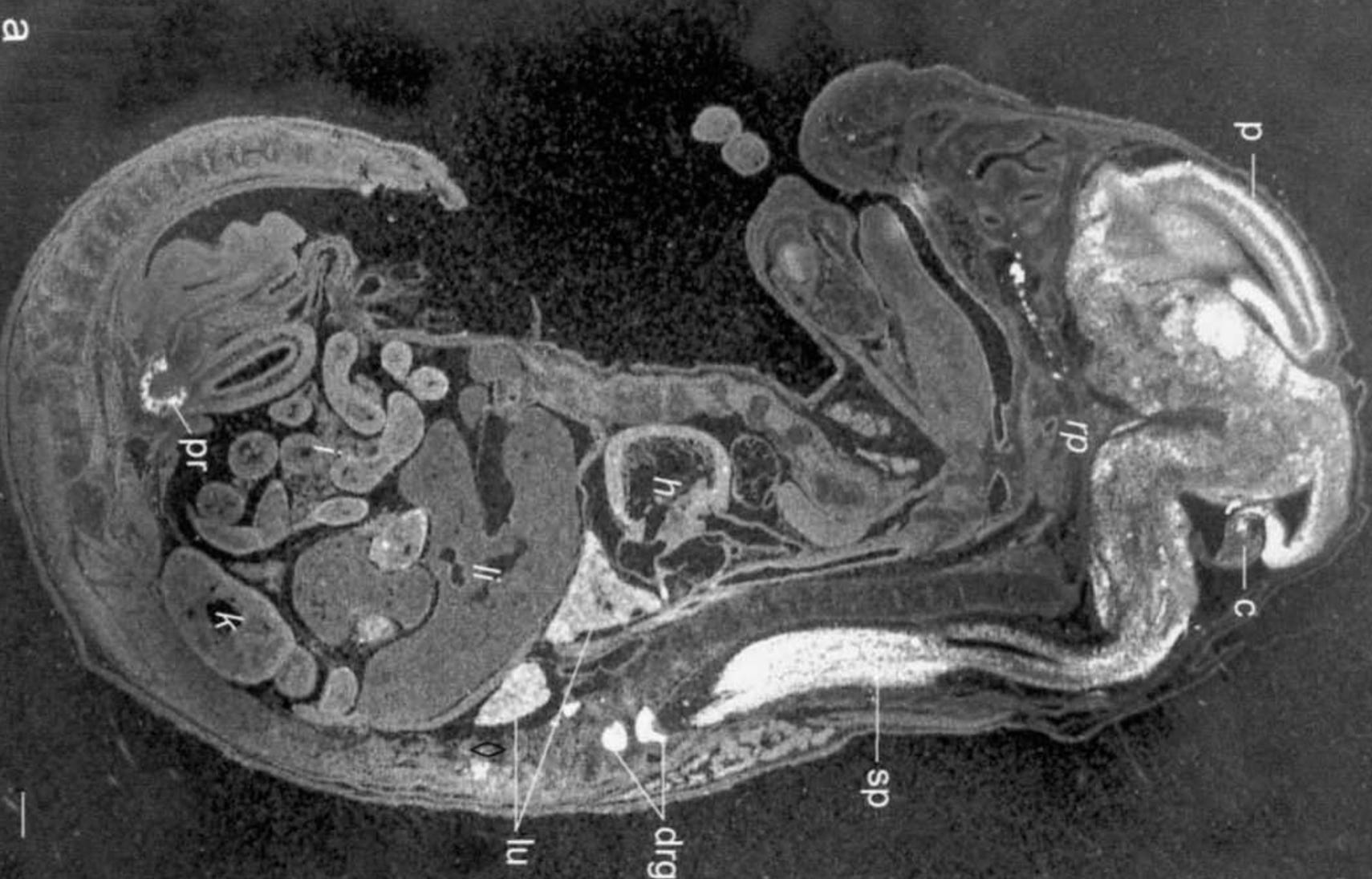
The authors started with a set of 319 individuals with HSV-1 encephalitis (HSE). They searched for mutations in genes previously known to be mutated in HSE, which yielded genetic diagnoses for 30 individuals. The authors suspected that the rest most likely are carrying mutations in genes that are not yet linked to HSE. There is a good rationale for that expectation.
Although primary herpes infections and secondary infections due to reactivation of latent virus are common, herpes virus spreading to the brain during reactivation is rare. It happens once or twice per 100,000 infections per year. And almost every such case is due to some defect in the brain-specific viral immune response pathways. When it comes to viral infections, brain is extraordinarily protected, thanks to evolution. You can appreciate this from the fact that even in conditions like primary immune deficiency caused by defective white blood cells production, the herpes infection do not spread to the neurons. Conversely, in individuals with HSE, rarely the infection spread to other parts of the body. Past genetic findings in HSE has reaffirmed the extraordinary cell-intrinsic nature of antiviral pathways.
Like we have seen in the cases of STAT1 deficiency, genes often has to be completely lost before the virus can infect the brain. Hence, the authors specifically searched for recessive mutations in the participants and found TMEFF1 in which homozygous coding variants were found in two individuals. The first was a French woman who had developed severe HSE at two and half years of age that left behind a large brain lesion visible in brain scan even 16 yrs later. The second was a 19 yr old Turkish man, born to consanguineous parents, who developed severe HSE at 5 years of age. Both recovered from HSE after intravenous acyclovir treatment, though not without neurological sequelae. Exome sequencing revealed that each carried a unique homozygous mutation in TMEFF1—a missense variant in the first and a splice variant in the second.
Given that TMEFF1 is a membrane protein, the authors hypothesized that its function has something to do with the virus entry into the neurons. The authors differentiated wild type and TMEFF1 knockout human pluripotent stem cell (hPSC) into neurons and infected them with HSV-1. As expected, the virus grew rapidly in the knockout cell lines but not in the wild types. The authors observed the same when using patient-derived hPSCs. The HSV-1 virus grew well in patient cells but not in the wild types with intact TMEFF1.
Next, the authors wanted to know if TMEFF1 acted through any of the known HSV-1 related pathways, for example, TLR3 and interferon alpha and beta signaling. It turned out TMEFF1 mechanism is unrelated to any of the known neuron-intrinsic antiviral pathways.
If the TMEFF1 acted at the membrane level, then it should be the first soldier to fight HSV-1 even before any of the others take out their swords. The authors created cell lines defective for known HSV-1 related immune genes like interferon receptor (IFNAR1) and tested if simply over expressing TMEFF1 would protect these susceptible cell lines from HSV-1 infection. It turned out, when over expressed, TMEFF1 confers protection even when key genes like IFNAR1 are inactive. More interestingly, the authors find that you don't even have to express the whole protein, just over expressing part of the protein that protrudes outside the membrane is sufficient to prevent HSV-1 from entering the cells.
HSV-1 like many other pathogens uses receptors to enter the cells. There are five glycoproteins (gB, gC, gD, gH and gL) and three receptor proteins (NECTIN-1, HVEM and PILRa) in the neurons that HSV-1 binds to to enter the cells. So, the TMEFF1 should somehow interfere with HSV-1's interaction with any these receptors to prevent its entry. Through co-immunoprecipitation experiments, the authors found a direct interaction between TMEFF1 and NECTIN-1. The authors further found that it is only the extracellular portion of TMEFF1 that interacts with NECTIN-1. So, the TMEFF1 protects neurons by interfering with HSV-1 binding to its receptor NECTIN1 to enter inside the cells.
What is the therapeutic implication of this discovery? Now we know that TMEFF1 has the ability to restrict HSV-1 entry into the cells, we can create synthetic soluble versions of this protein and use it like a vaccine to prevent HSV1 infection or reactivation. Although the present findings support a therapeutic role for TMEFF1 in HSV1 infection, the authors expect that the approach would be also work in HSV2 infection.
The main take home for me from this study is that biological diversity between cell types in humans offers an unique opportunity to mine targets. In this case, the fact that brain cells have evolved their own antiviral defense mechanisms opens door to a new class of evolution-validated drug designs. The idea aligns closely with our previous story of Big tau expressed only in brain tissues resistant to neurodegeneration in Alzheimer's disease.

I am excited about this emerging new idea of leveraging within individual biological diversity to uncover novel drug targets. I am hoping to see more such breakthroughs in the future.

After reading, I'm really curious what I may have going on infection-related genetics. I have had a lot of HSV-1 exposure but have never been infected (or at least never have symptoms of infection). Contrastingly, I struggle with clearing plantar wart infections. This is gross but when I was in my early teens I have dozens of plantar warts on my feat that only candida injections cleared.
Splendid narration….. its sherlockian who done unit writing keeps the reader engrossed…..complex exciting research and its potential implications explained succinctly and simply. Kudos to you
Would look forward to your insights on miRNas ,especially miRna34 in hepb hepc and fibrosis in liver in following write ups