
The Big tau: A new hope for Alzheimer's?
An overlooked tau isoform may hold the key to an effective Alzheimer's drug


I love coincidences, particularly the ones related to scientific discoveries. In 1975, particle physicists at the Stanford University and University of California in the US published a discovery of a new elementary particle. Twenty years later, the discovery would fetch Martin Perl, the lead physicist, a Nobel Prize in Physics. Following the ancient tradition of naming things using Greek letters, the newly found particle was named "tau"(τ).
In the same year, scientists from the Princeton University in the US published a discovery of a mystery protein in the brain tissue of pigs. Like how bones form the skeleton of the human body, there are protein complexes--microfilaments, microtubules and intermediate filaments--that form the skeleton of our body cells. Microtubules are long tubular structures built from tiny lego-like pieces called tubulins. There are two types of tubulins: alpha and beta. The two combines to form heterodimers. The heterodimers further assemble into microtubules.
When you crush a microtubules into pieces, you find among the pieces individual tubulin dimers, and also, pieces of ring-like structures each consisting 23 tubulin dimers (which can be further broken down to individual dimers). When you try to polymerize the broken pieces, the larger rings easily come together and transform into microtubules. But the individual dimers don't. Some mysterious factor seems to hold the tubulins together and help them to polymerize. Marc Kirschner and colleagues from Princeton discovered that mystery factor to be a protein. Honoring the ancient tradition of naming things using Greek letters, the newly found protein was named "tau"(τ).
In previous studies of in vitro microtubule assembly, it was shown that depolymerized tubules contain two components: the 6S tubulin dimer, and a 36S species which contains about 23 tubulin 6S subunits arranged mainly as double rings. We observed that only the fractions containing 36S rings would repolymerize to form microtubules; the 6S dimer alone would not. ...
We now report that the ability of 36S rings to assemble into microtubules is due to a salt dissociable factor which resides in the 36S species but is absent in the 6S dimer. We have isolated this factor, a protein which is essential for the assembly of 6S tubulin into 36S rings and microtubules. We propose to call this protein tau (τ) for its ability to induce tubule formation.
Both the physics tau and biological tau represents major discoveries in their respective fields. But it was a mere coincidence that they happened to be born in the same year and to be given the same name. If you Google "tau", the first result will be a Wikipedia link to the physics tau. You need to search for "tau protein" to read about the biological tau.

Tau is encoded by MAPT (microtuble associated protein tau) located on chromosome 17. It is highly expressed in neurons where it helps to stabilize the microtubules. Neurons need microtubules not only to maintain their structures but also to communicate with each other. You'd think that tau sounds like a very important protein and without this protein organisms will not survive. Surprisingly, it’s not true. You can delete the MAPT gene from the mice safely without any major consequence, and the mice still will live and breed normally. It was one of the surprising revelations in the Alzheimer's research (Ke et al. Int J Alzheimers Dis 2012.)
Then why does this protein wreak havoc in the brains of individuals with conditions such as Alzheimer's disease, frontotemporal dementia, progressive supranuclear palsy (PSP) and Pick's disease, etc.? Tau proteins are complex proteins with many structural domains that make them inherently unstable. Many factors can destabilize tau and make them stick together to form tau tangles. Mutations that result in abnormal secondary and tertiary structures (misfolded tau proteins) can lead to sticky tau. Certain splicing mutations add or remove certain parts of the tau protein and consequently, increase and decrease the sticky tendency. The tau proteins have certain amino acids like serine and threonine in their structure that can accept a phosphate group. Microtubules are dynamic structures. Correctly adding and removing phosphate groups to and from the tau protein, respectively, is essential for the tau to assemble and dismantle the microtubules. Too many phosphates can kick the tau off the microtubules, resulting in tau aggregation. A worst thing about tau aggregation is it can propagate. As one tau protein start sticking to an another, the next and next tau proteins follow suit and the chaos can gradually spread across the brain like it occurs in Prion diseases.
One interesting feature of the MAPT gene is that its nascent mRNA transcript are spliced in different ways to producing a variety of isoforms that subtly differ from each other. In the context of central nervous system, splicing of three three exons (2, 3 and 10) determines the isoforms. Based on presence or absence of exons 2 and 3, the protein is labelled 0N (neither exons are included), 1N (only exon 2 included) and 2N (both exons 2 and 3 are included). The exon 10 codes for a repeat region that forms the microtubule-binding interface. Based on the presence or absence of exon 10, the protein is labelled as 4R (four repeats) or 3R (three repeats)1. So, alternative splicing of the three exons (2, 3 and 10) gives rise to six different isoforms: 0N3R, 0N4R, 1N3R, 1N4R, 2N3R, 2N4R.
While the six isoforms are mainly found in the central nervous system (CNS), in early 1980s, scientists came across a new high molecular form of tau protein in the peripheral nervous system (PNS) tissues like spinal cord, spinal ganglia, peripheral nerves etc. This larger isoform was named "big tau" or "PNS tau". The exon contributing to the extra length of big tau was cloned in the early 1990s (Goedert et al., Couchie et al. PNAS 1992). As the MAPT exons were already numbered by then, the newly found exon located between exons 4 and 5 was given the number 4A (shown as black rectangle in the figure below).
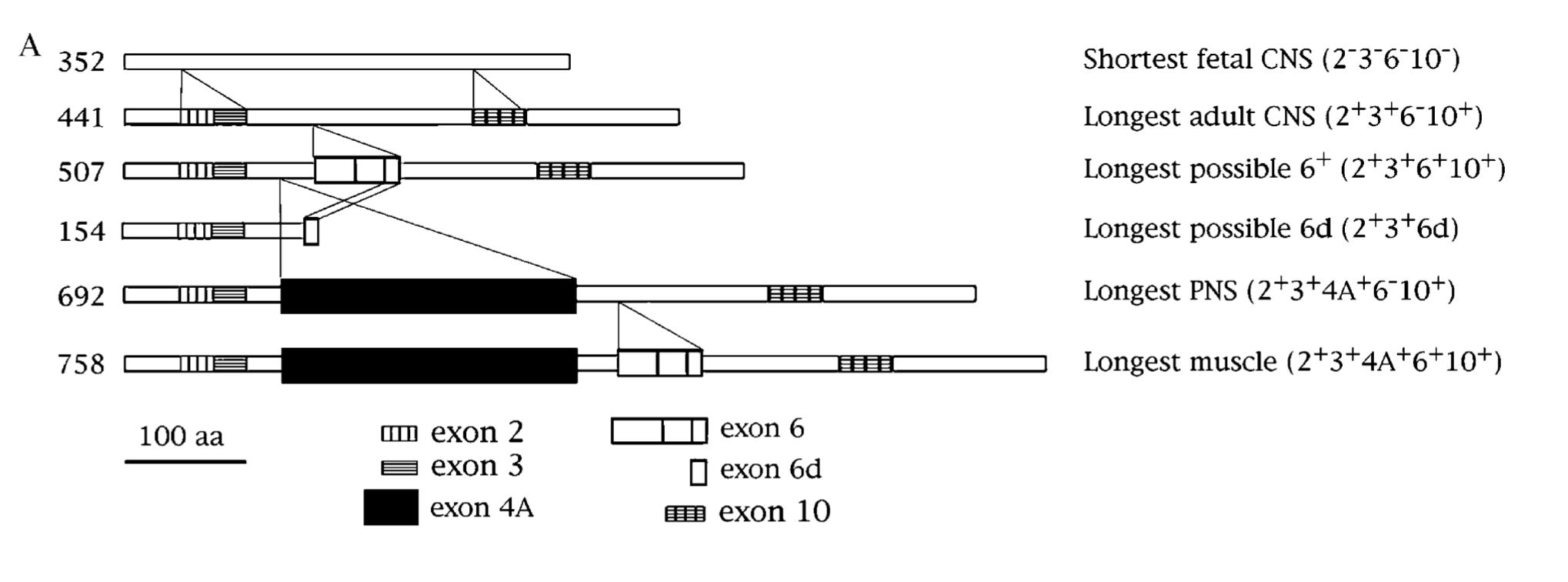
For some reason, the big tau never attracted much attention from neuroscientists for more than 25 years after its discovery (Fisher eNeuro 2023). Now the big tau is making a comeback. A new study from the lab of Huda Zoghbi, a world-renowned neuroscientist known for her pioneering work in conditions like Rett syndrome, spinocerebellar ataxia 1 etc., reveals the extraordinary qualities of big tau, nominating it as a potential therapeutic target for tauopathies.

In the preprint, Chloe Chung et al. reports that, on contrary to the popular belief, the big tau is expressed in the brain. The authors show this both in mice and humans. It turns out the brain regions expressing the big tau are the ones that are typically spared from neurodegeneration in the Alzheimer's disease. Using western blot, the authors show that big tau is abundantly expressed in the mouse cerebellum and brain stem. The authors further show the same in the postmortem brains of Alzheimer's patients and controls. Specifically, the authors show the big tau is more abundant in the cerebellum of Alzheimer's brain than that of controls.

Motivated by the abundant expression of big tau in brain and its unique structure with an N terminal end that is three times longer than the regular tau isoforms, the authors studied the biochemical properties of big tau. As I mentioned before, one of the key chemical properties of tau is phosphorylation. The level of tau phosphorylation increases with age, and it also increases in the diseased state. By measuring the phosphorylation levels of big tau in younger and older mice, the authors show that big tau is less subject to age-associated phosphorylation.

One of the ways through which the neurons clear the tangled tau proteins is ubiquitination, a process of adding little protein tags to signal the cellular protein clearance system. The ubiquitin are added to lysine residues, and it turns out the big tau contains 20 additional lysine residues than the regular tau. The authors show that the big tau gets more easily ubiquitinated and consequently, gets efficiently cleared, compared to the regular tau. And this property makes lifespan of big tau much shorter than the regular tau.
One of the main problems with tau proteins in Alzheimer's disease is they easily fall off the microtubules. When you centrifuge the microtubules at high speed, the tau that are not tightly bound to microtubule get separated, which can be collected as a supernatant. The authors show that, unlike regular tau, the big tau doesn't get separated from microtubules. After ultra-centrifugation, they are still found bound to microtubules at the bottom. What makes the big tau bind more tightly with microtubules? Using a combination of bioinformatics and microtubule binding assay, the authors show that big tau possess extra microtubule binding motifs. The binding affinity is so strong, in a competitive binding assay, the big tau outcompetes regular tau even when present in small amounts.

Finally, the authors test the most critical property of the tau proteins—aggregation tendency. There are certain mutations that increase the aggregation tendency of tau proteins. The authors show that even if you introduce such a mutation, the big tau refuses to aggregate, both in vitro and in vivo (in the mouse brain).

Overall, the authors demonstrate that big tau has many properties that make it resistant to Alzheimer's disease pathology: it resists hyperphosphorylation, it gets easily cleared, it binds tightly to microtubules and it refuses to aggregate, even in the presence of aggregation-promoting mutation. How amazing is that? I am surprised that this protein has evaded scientists' attention for nearly 3 decades since its discovery.
The discovery has a major therapeutic implication. Tau is an important drug target of interest for many companies. Many modalities are being tested. Many are developing antibodies to bind and clear the tau tangles. Some are developing small molecules to prevent tau aggregation. Some are developing siRNAs to knock down MAPT expression itself. The new findings opens a new therapeutic idea. If big tau is resistant to aggregation, then one can try a non-destructive way of treating tauopathies, for example, instead of completely shutting off the tau production, one can simply nudge the neurons using splice modulators to produce big tau instead of regular ones. Its not as easy as it sounds, but definitely worth trying. I am sure many companies will do.

One thing that came to my mind when reading this paper was the recent Casgevy approval for the treatment of sickle cell disease2. Casgevy is a CRISPR-based treatment. The patients' blood stem cells are taken out of the body, gene edited to reduce the expression of a gene called BCL11A and then put back into the patients. The BCL11A is a key regulator of fetal to adult hemoglobin switching. By reducing the BCL11A expression, Casgevy increases the fetal hemoglobin production, protecting the red blood cells from sickling and thereby, increasing the quality of life of the patients. I wonder if there exists any splicing regulatory protein that can be targeted to increase the big tau production in the neurons.
One key take away from this work for me is, we human geneticists always go in search for genetic diversity across humans to find drug targets. But there is incredible genetic diversity within individual human brains that has been largely unexplored to date. The current work from Huda's lab and the recent work on Huntington's disease from Steve McCarroll's lab are great examples. I expect we will see more such work in the near future.

Most frontotemporal dementia (FTD) causing mutations cluster around exon 10 (Wang et al. J Neurochem 2004)
I’ve written about Casgevy in a previous Substack post and discussed about it with Patrick Short in an episode of The Genetics Podcast.
Are there tau knockouts in human population? Or tau aggregation resistant individuals? If so, do we have any close phenotyping of such individuals?
So the reason why MAPT-KO mice exhibit normal phenotype is still uncovered?