
De novo enhancer creation by a noncoding mutation
Genetic investigation of a cardiac arrhythmia reveals a new noncoding Mendelian disease mechanism


Decoding the noncoding genome has been a recurring theme in my past posts. Today, I have another fascinating story under this theme. It's about a noncoding variant underlying a newly discovered rare, Mendelian heart condition characterized by a distinct ECG pattern and sudden cardiac death.
One of the biggest mysteries of human genome that we will see scientists incrementally solve over the next decades is the biological mechanism through which noncoding variants influence disease-risks and trait variations. Solving this mystery not only will bring genetic diagnoses for hundreds of rare diseases (as we've seen in the recent RNU4-2 story) but I believe it will also bring breakthroughs in drug development.
As a scientist working in drug development, I try to understand how genetic variants increase or decrease gene function, and how this information could be used identify potential drug targets. So far, the field has been mainly relying on coding variants to do this. We specifically look for rare coding variants to understand the beneficial and harmful phenotypic consequences of increasing or decreasing the function of a gene.
There are limitations to relying solely on coding variants. For example, most of the variants often decrease the gene function rather than increase it. Of course, there are examples of coding variants that increase the gene function. But they are not as common as loss of function variants, and also, they are neither easily identified nor confidently interpreted. The ones we confidently interpret today are mostly loss of function variants such as frameshift, stop gain variants that truncates the protein and abolish its function.
Another limitation of studying coding variants is that their effects are often omnipresent in the human body across space and time. A person born with one copy of a protein truncating variant in, let's say, gene X will be deficient of that gene since their conception in utero until their death. They will be deficient of the gene in every cell of their body that expresses it and at every phase of their life when the gene is expressed. Of course there are exceptions like certain splicing variants affecting specific transcripts that show tissue-specific expression, but such examples are rare.
Noncoding variants are expected to address many of the limitations of coding variants like the ones I mentioned above, but we are at our infancy in understanding of how sequence changes in the noncoding genome impact functions of neighborhood or distant genes. Our knowledge are slowly evolving with emerging discoveries, particularly the ones made in individuals with rare Mendelian diseases.
Gene often express tissue and development stage specifically. These space and time restrictions are coded in the regulatory elements widespread across the noncoding genome. Mutations within such regulatory elements can cause severe consequences, sometimes more severe than coding mutations, and through discovery of such mutations, scientists learn about the existence of critical noncoding regulatory elements and the mechanisms through they control their target gene expression.
I've highlighted in my past posts many examples of discovery of noncoding variants underlying Mendelian diseases. One of my favorites is the discovery of intronic variants in HK1 that derepress the hexokinase expression in pancreatic beta cells, resulting in uncontrolled insulin secretion and fatal hypoglycemia in infants with congenital hyperinsulinism. Here, the noncoding variants taught scientists about the consequence of expressing hexokinase in beta cells, where they are not supposed to express.

Another example is the discovery of a noncoding structural variant underlying a monogenic form of extreme obesity that rewires the gene expression program of ASIP, resulting in ubiquitous expression of ASIP instead of skin-specific expression. Here, the noncoding variant taught scientists about the consequence of expressing ASIP in brain hypothalamic neurons where they are not supposed to express.

Another example is the discovery of a noncoding variant underlying a neurodevelopmental disorder that abolishes the expression of a long noncoding RNA and as a result, amplifies the expression of nearby neurodevelopmental gene CHD2.
Among the noncoding variants, the ones that cause gain of function effect is of particular interest for me for the reason that I explained before. From past examples, we have learned that noncoding variants can achieve gain of function effect through duplication of a regulatory element (as was the case with ASIP), loss of inhibition of gene repression (as were the cases with HK1 and CHD2). In a recent preprint, a research team from University of Oxford report a new mechanism through which noncoding variant causes a gain of function effect: de novo creation of a cardiomyocyte-specific enhancer.
The story starts 36 years ago when cardiologists at the Copenhagen University Hospital in Denmark encountered a 30 years old patient who incidentally presented with a puzzling ECG feature: persistent ST segment depression1 without any associated cardiac disease. The patient appeared healthy. The doctors followed this index patient for the next 30 years. After remaining asymptomatic for 25 years, the patient developed atrial fibrillation at 55 years of age. Eight years later, he developed ventricular fibrillation and was rescued from sudden cardiac death. Both his children, the doctors found, had similar ECG changes as his. The doctors learned that two of their family relatives have died in the past from sudden cardiac death. While studying this index family, the doctors also encountered a series of other families with similar history of atrial and ventricular arrhythmias associated with ECG changes strikingly similar to that of the index family. They realized that are dealing with a new, genetic cardiac syndrome, which is passed down in the families in an autosomal dominant fashion. They published the case reports in NEJM in 2018.
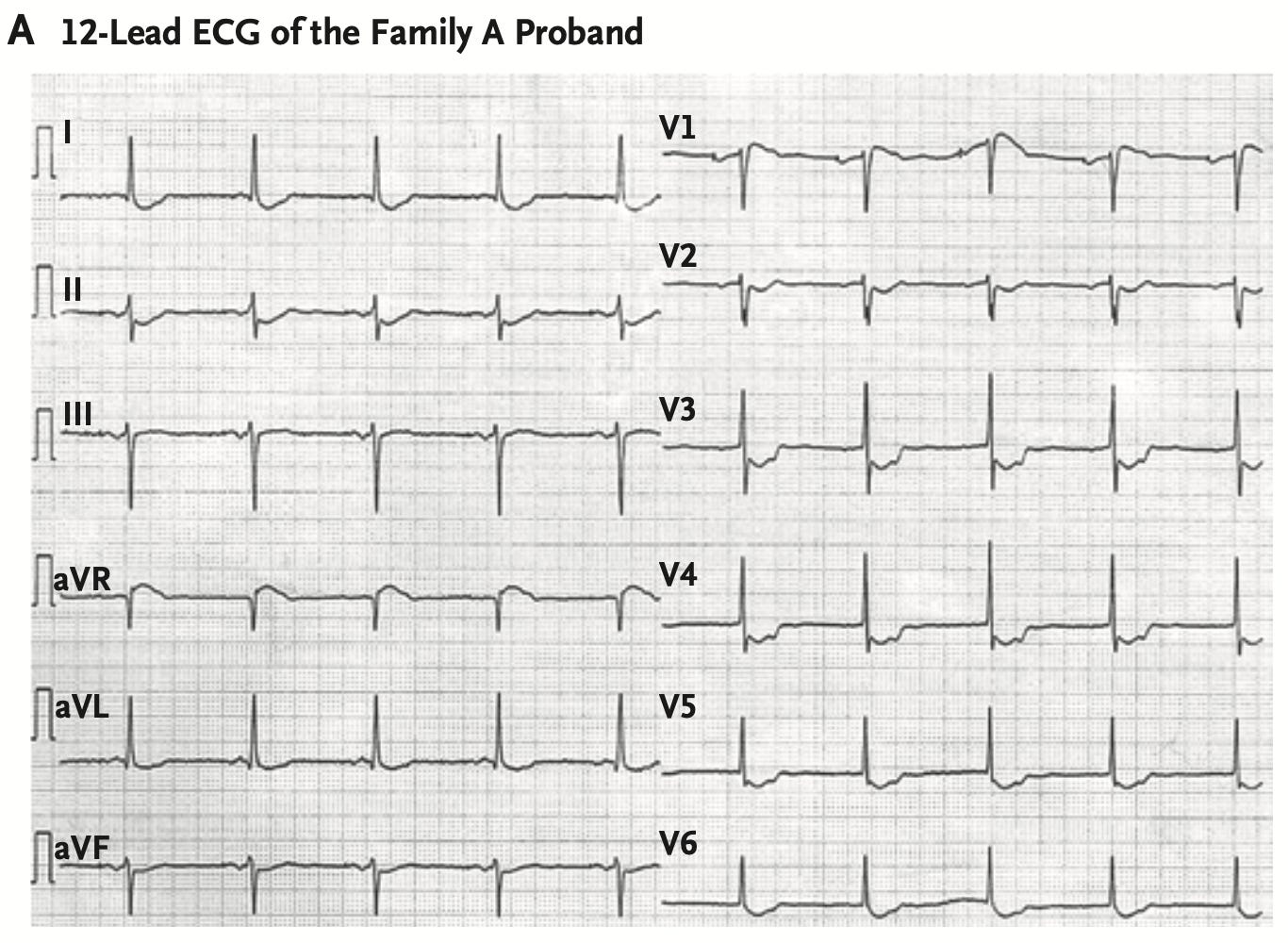
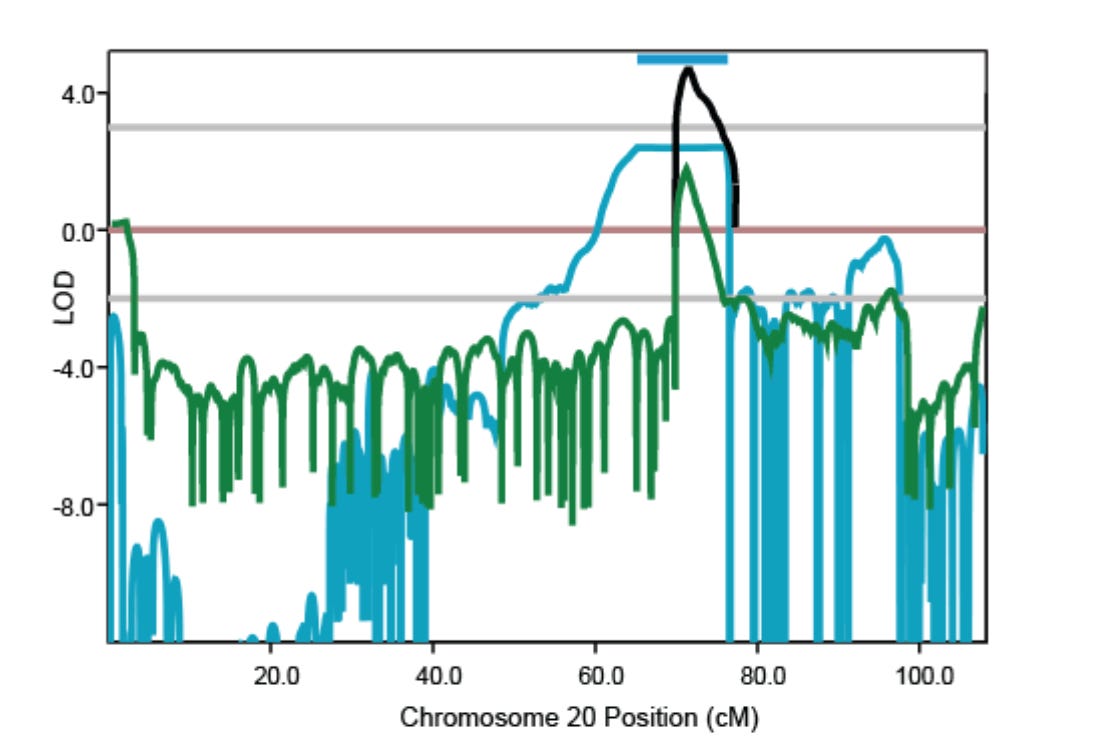
The current preprint is a genetic follow up of the cardiac syndrome, named as ST depression syndrome (STDS), reported in 2018. Using linkage analysis of the affected families, the authors mapped a locus in chromosome 20 that was shared by two different families. Through further analysis of sequence variants within the chromosome 20 locus, the authors identified the disease haplotype, which turned out to be same in both the families, and also, in another patient from a third family. The authors suspect that all the affected families are distantly related and have inherited the disease variant from a common founder. Through further analysis, the authors narrowed down the disease variants to a single heterozygous noncoding variant that fell within the linkage region and was found in every affected family member but not in the unaffected. It was a complex deletion-insertion variant characterized by loss of 17 and gain of four nucleotides. The authors call this variant as "delinsTCCC".
My favorite part of the story is how the authors followed up this variant further to arrive at the causal gene and the variant mechanism. The region containing the delinsTCCC variant turned out to be highly conserved and not that many genes were in the vicinity. The nearest one was ~7kb away, KCNB1, encoding a potassium channel. Studying previously reported GWAS signals at this location, the authors found that there was indeed a GWAS signal nearby, mapped to ECG ST segment changes. The delinsTCCC variant was sitting in between two index variants of the GWAS locus. Analyzing low-resolution chromatin conformation capture data from cardiomyocytes differentiated from induced pluripotent stem cells (iPSCs), the authors found that one of the GWAS variant and delinsTCCC fell within a region that physically interacts with the promoter of KCNB1.
The authors further analyzed epigenetic data based on relevant tissues and cell types from external sources and found that the delinsTCCC did not overlap with any open chromatin region in cardiac cells and the nearest open chromatin region was 12 kbp upstream, sitting between delinsTCCC and the KCNB1 promoter. The authors call this regulatory element as "E-139" (as it was 139 kbp away from the KCNB1 promoter). The authors next studied the expression profile of the E-139 region and found that it is expressed in skeletal muscle and many cancer cell lines, but puzzlingly not in the cardiac muscle. The authors had an intuition that delinsTCCC might have created a new enhancer-like element. As they suspected, a neural network-based sequence prediction model informed that the delinsTCCC sequence indeed have features suggestive of open chromatin, accessible to cardiac-related transcription factors. The authors gene edited iPSC cells to introduce the delinsTCCC variant and differentiated them into cardiomyocytes, and using ATAC seq, they confirmed that the delinsTCCC sequence opens up the chromatin and binds to enhancer-related histone proteins.
The authors went one step further and created transgenic zebrafish lines with the delinsTCCC variant or the nearby E-139 element or corresponding wild type sequence, all placed near a GFP protein with a minimal promoter. As expected, the delinsTCCC model showed fluorescence specifically in the heart cells, confirming that the mutation sequence acts as an cardiac-specific enhancer.

Surprisingly, the E-139 model showed florescence ubiquitously (only mildly in the heart), particularly in the brain. This aligned well with the known neurodevelopmental consequence2 of KCNB1 coding variants. It turns out the KCNB1 potassium channel plays a role in brain development, and probably, has no business in the heart. But the mutation made these potassium channels to express in the heart, resulting in cardiac conduction defects.
Finally, the authors performed chromatin capture assay in gene-edited cardiac cells and tested if the delinsTCCC physically interact with KCNB1 promoter. This is where they found something interesting. It turned out that, unlike E-139 element, the delinsTCCC sequence element doesn't physically interact with the KCNB1 promoter (the physical interaction they initially observed was based on low resolution Hi-C; here they are looking at high-resolution). But interestingly, the physical interaction between E-139 element and KCNB1 promoter became amplified in the presence of delinsTCCC. The findings suggest that delinsTCCC region might not be a conventional enhancer, but might function as a super enhancer that facilitates the interaction between nearby enhancer (E-139) and promoter (KCNB1).

This study is one of the many examples to remind us that we should reconsider our notions about disease-causing variants. Conventional thinking would be to search for variants in regions that are previously annotated as promoter or enhancer in disease-relevant tissues. The study reminds us that disease-variants not only can disrupt already existing regulatory elements but it can also introduce an entirely new regulatory element. Similarly, when prioritizing disease-genes, it is a common practice to narrow down the list by excluding genes that not expressed in the disease-relevant tissues. The study reminds us that disease-variants not only can affect genes that are specifically expressed in disease-relevant tissues but it can also affect genes that are specifically not expressed in disease-relevant tissues.
The authors write "we believe this is the first description of an entirely de novo cryptic enhancer causing a Mendelian disorder", that such type of examples may be more common that one would expect, and with increasing use of whole genome sequencing to diagnose rare diseases, we will learn more of similar noncoding mechanisms in the future.

Since recently we constantly hear about Indian actors, men around 40 succumbing to cardiac arrest. These cases must be more common. What do you believe is the cause?
Is there an existing database or catalog of non-coding variants associated mendelian disease? Is there an easy way to search OMIM for these (if they're even in OMIM)?