
Nature's undoing of the curse of Colombian Alzheimer's families
Genetic modifiers of Alzheimer's disease-causing mutations

Last week, I was recording with Patrick the Q2 podcast episode of human genetics round up. For those who are not familiar with the series, in the end of 2022, I spoke about some of the interesting human genetics papers published that year on The Genetics Podcast hosted by Patrick Short from Sano Genetics. Motivated by the positive feedback, Patrick asked me to do it again in 2023, and then it became a routine. This year, we've planned for four episodes, one each quarter. I've always struggled to decide which ones to discuss, as always there were more great papers than we could cover in one hour. But somehow I manage to pick a few interesting ones, of course, based on what I read recently and what stayed in my memory. The Q2 episode will be out in a few weeks from now. One of the papers I discussed was the recent Alzheimer's disease (AD)-related one published in NEJM. It was about a rare APOE Christchurch mutation as a potential genetic modifier of PSEN1 E280A mutation that causes a severe early onset form of AD.
PSEN1 encodes presenelin 1, a component of a large multiprotein enzyme complex called gamma-secretase. This enzyme complex is what cleaves the amyloid precursor protein (APP) resulting in amyloid beta peptides that aggregate to form the amyloid plaques, seen in the brains of AD patients. Certain missense mutations in PSEN1 cause a severe form of autosomal dominant AD with very early onset, discovered in the mid 1990s. The largest of AD families affected by PSEN1 mutations resides in Antioquia in Colombia. It's a large pedigree of nearly 6000 members of which around 1000 individuals carry a pathogenic missense mutation, E280A1, in PSEN1. The Antioquia Alzhiemer's family has attracted a great deal of interest from AD researchers. Nowhere else in the world one can find so many AD patients, all caused by a single mutation. So, naturally, scientists have been closely watching this kindred to identify any outliers. In 2019, researchers from Harvard Medical School and Universidad de Antioquia (Arboleda-Velasquez, Lopera, O’Hare, et al. Nat Med) reported one such outlier: a Colombian woman with E280A mutation who was destined to develop Alzheimer’s in her late 40s, but never did until her 70s. It then turned out this woman was gifted with a rare APOE mutation2 called Christchurch mutation in the homozygous state. The Christchurch mutation reduces the APOE function3. It was the first ever genetic evidence to support the concept of targeting APOE protein to treat Alzheimer's disease in the absence of APOE4 mutation.
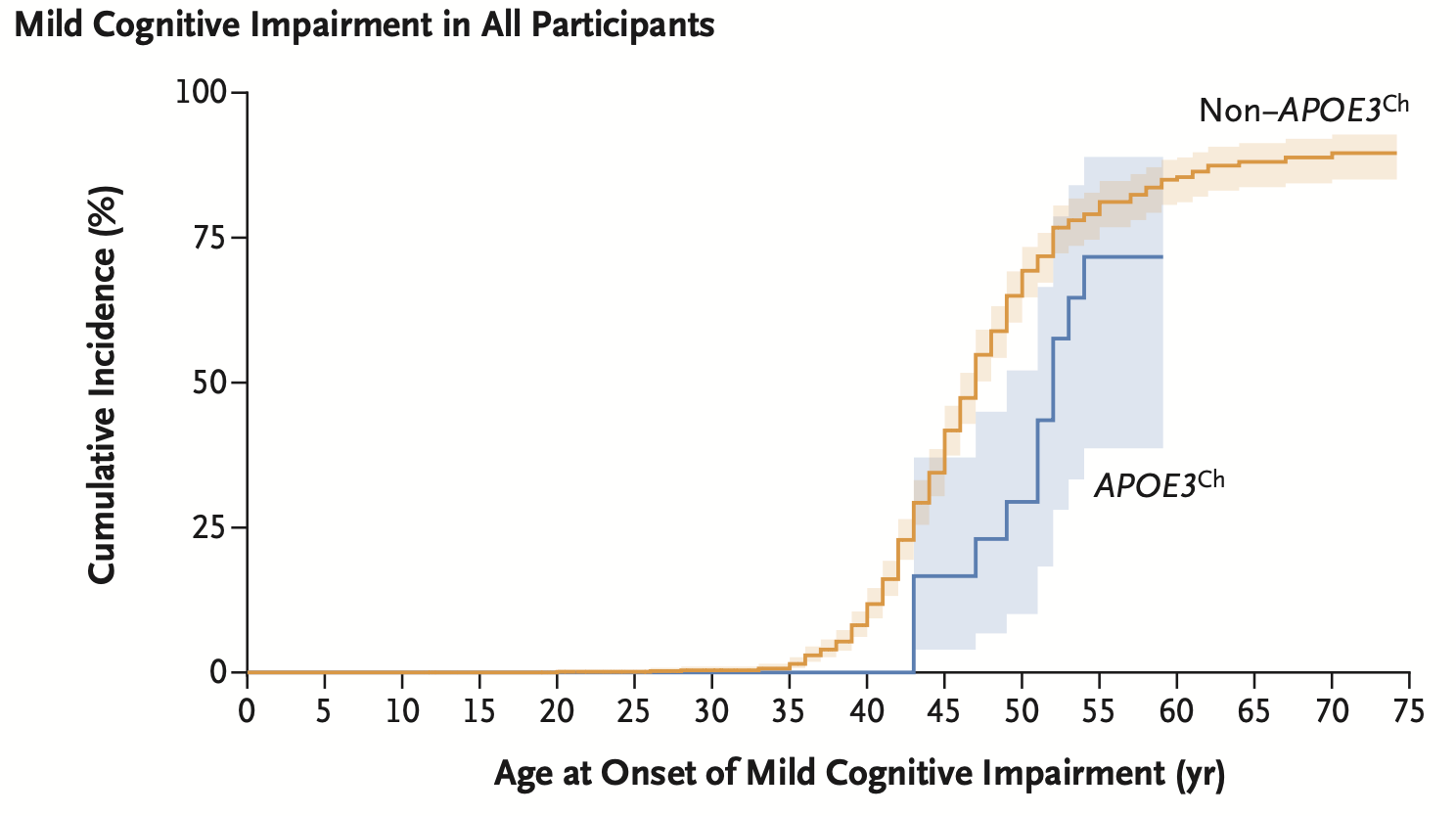
Although APOE has always been a therapeutic target of interest, the discovery of the Christchurch mutation dampening the PSEN1 mutation effect hinted that the target population for an APOE-targeted medicine could involve more than APOE4 carriers. However, that was just an n=1 study. The authors wanted to know if this protective effect is seen even in heterozygous carriers of Christchurch mutation. So, they screened the Antioquia pedigree and found 27 carriers. They studied retrospectively the longitudinal clinical data of these individuals, including the autopsy reports of four deceased individuals. The authors (Quiroz, Aguirre-Acevedo, Vasquez, et al. NEJM) have reported their observations in NEJM recently. The bottom-line is that E280A carriers who also carried one copy of Christchurch mutation appear to have a relatively less severe Alzheimer's disease compared to other E280A carriers. The age of onset of cognitive impairment was delayed by around 5 years. Brain imaging and autopsy examination revealed limited tau pathology4 and neurodegeneration.
One of the interesting findings in both the earlier and current reports is the presence of high amyloid beta plaques in the brain despite a limited tau pathology. The postmortem brain examinations of both the homozygous woman in the earlier report and four deceased heterozygous carriers in the current report show the brains of these individuals had high amyloid beta plaques comparable to that of other E280A mutation carriers. So, the protective effect is seen despite high amyloid beta burden, which hints that APOE targeting might work even after the patients start making high amyloid beta plaques in their brain. The finding also adds to the long-running debates on amyloid hypothesis in AD, perhaps hinting that the AD treatment approaches shouldn't be exclusively focussed on clearing amyloid deposits in the brain.
The APOE mutation modifying the penetrance of PSEN1 mutation is an example of trans-genetic modifier. That is, the modifier (APOE) and modifiee (PSEN1) genes are located on different chromosomes. Recently, there have been also reports of cis-genetic modifiers in AD. In the last year round up, I highlighted a preprint by Michael Greicius and colleagues from Stanford University on APOE loss of function mutations eliminating the pathogenic APOE4 alleles. The paper was published in Neuron a few months ago. It's a nice example of cis-genetic modifier.
APOE4 is the most common genetic risk factor of AD, with E4 homozygosity is now widely recognized as a highly penetrant, monogenic cause of AD. One of the therapeutic approaches drug developers are trying is to treat AD (caused by APOE4 mutation) by inhibiting APOE. Hence, there is interest in studying the effect of APOE loss of function mutations in the population. However, individuals with loss of function mutations in APOE are rare. The Stanford researchers went one step further and looked for an even rarer set of individuals: those with both APOE loss of function mutation and APOE4 mutation in the same chromosome. Surprisingly, they did manage to find a handful of such individuals by scanning through an AD cohort. The authors found that individuals who carried APOE loss of function mutation in the same chromosome as E4 allele did not develop Alzheimer's until their old age and did not show evidence of Alzheimer's pathology in the brain images or in the CSF. The major limitation of this paper is there were only a handful of carriers, and none of them were E4 homozygotes. Since E4 heterozygosity is not that highly penetrant as E4 homozygosity, it's critical to demonstrate protective effect in homozygotes. Given now there is a consensus that E4 homozygosity is near 100% penetrant, identifying even one individual with E4 homozygous genotype and a APOE4 loss of function variant and showing that the AD severity is reduced in this individual will strengthen the case for APOE-targeted therapeutics for AD.
Genetic modifier is an emerging topic in the drug development field lately. In fact, one of the main goals of drug companies investing in human genetics is to identify genetic modifiers that can validate their drug designs. With the rapidly growing genetic databases, interesting examples are beginning to emerge. I’ve discussed one such example in my earlier posts: a loss of function missense variant, N264K, in APOL1, abolishing the effect of kidney disease-causing APOL1 mutation5, which validated the therapeutic approach of inhibiting APOL1 to treat chronic kidney disease.
Another disease area where genetic modifiers are a hot topic is repeat mutations-associated neurodegeneration like Huntington’s disease, cerebellar ataxia, amyotrophic lateral sclerosis (ALS) etc. Our ability to investigate the genetic modifiers of repeat mutations in the population so far has been limited due to unavailability of whole genome sequencing datasets, as often the disease-causing repeat mutations are located in the noncoding genome. As a result, search for modifier variants focussed heavily on common variants. For example, a genome-wide association study of age of onset of Huntington’s disease has reported a common variant near MSH3 that encodes a DNA mismatch repair protein. The finding aligns with the emerging interest on targeting mismatch repair genes to prevent somatic repeat expansion of Huntington’s mutation. Large-scale sequencing projects focussed on Huntington’s disease are currently in progress. Hopefully, such efforts will reveal more genetic modifiers of therapeutic value.
One of the limitation of studying genetic modifiers, particularly in the disease areas of neurodegeneration, is lack of good endophenotypes. In the case of APOL1, the researchers have a great surrogate marker which is in vitro trypanolytic activity. Another example where availability of a good enophenotype accelerated drug target discovery is sickle cell disease (SCD). The BCL11A was discovered through studying the genetics of blood fetal hemoglobin fraction, which is a strong causal predictor of disease progression in SCD. Remember, SCD is a monogenic condition caused by a single point mutation in the HBB gene. So, the common variants near BCL11A discovered via GWAS are in fact trans-genetic modifiers of HBB mutation. If it were the case that the researches had to discover genetic modifiers of HBB mutation only by studying by correlating genetics with clinical markers of disease progression of SCD, we wouldn’t have got an FDA-approved CRISPR-medicine for SCD today. Speaking of which, scientists are already in track to developing small molecule drugs that can up regulate fetal hemoglobin in the blood to treat SCD. Although there are concerns of off-target effects, it’s a big breakthrough of the year as it opens the possibility of treating SCD using cheap oral pills rather than expensive CRISPR therapy.
To conclude, I expect many discoveries of genetic modifiers of disease causing mutations in the near future. These discoveries will be driven by large-scale sequencing efforts, natural history studies of disease mutation carriers, and importantly, innovation in phenotyping methods.

Amino acid change from glutamic acid to alanine at position 280 in the PSEN1 protein.
Amino acid change from arginine to serine at position 136 in the APOE protein.
The different APOE variants in decreasing order of pathogenicity: E4> E3 > E2 & Christchurch
Tau is a neuronal cytoskeletal protein that becomes abnormally phosphorylated and aggregate to form neurofibrillary tangles. It’s a marker of neurodegeneration and can be measured in brain through PET imaging and in the CSF through biochemical assays.
So in behaviour genetics, you often hear that genetic effects are mostly linear with few interactions. But in medicine, genetic modifiers are being studied, ie interaction effects?
Also, a piece of jargon I didn’t get, what’s penetrancy?